DOI:
10.1039/C6RA05990B
(Paper)
RSC Adv., 2016,
6, 44641-44645
(Fe1−xNix)3N nanoparticles: the structure, magnetic and photocatalytic properties for water splitting†
Received
7th March 2016
, Accepted 14th April 2016
First published on 21st April 2016
Abstract
Fe3N and (Fe1−xNix)3N nanoparticles (NPs) were prepared via a simple sol–gel method. The structures, morphology, and magnetic properties of the NPs are investigated using X-ray diffraction, X-ray photoelectron spectroscopy, transmission electron microscope. It is shown that (Fe1−xNix)3N possess a spherical morphology with a core–shell structure from the TEM results. The magnetic properties of Fe3N and (Fe1−xNix)3N NPs are carefully studied by using vibrating sample magnetometer. Moreover, Fe3N and all of the (Fe1−xNix)3N NPs are found to photocatalyze water splitting under Xe lamp irradiation. These results suggest that (Fe1−xNix)3N NPs can be used as a low-cost catalyst for water splitting.
Introduction
Recently, the application of semiconductors for photocatalytic H2 production from water has attracted tremendous interest.1 Many studies have indicated that semiconductor photocatalysts containing noble metals (such as Pt, Au and Ag) display high photocatalytic activity.2–6 However, the scarcity and high cost of the noble metals have greatly hindered their application. Despite the many oxide/sulfide photocatalysts that have been developed for this application in previous research,7–9 the performance of these catalysts was greatly restricted by their poor chemical stability. Recent research about semiconductor photocatalytic materials has focused on metal nitrides.10–13 Among these nitrides, iron nitrides, especially ε-Fe3N, have gained particularly considerable interest due to their expedient magnetic properties, stable chemical properties and good thermal stability. It has previously been shown that ε-Fe3N could be used as a catalyst to produce hydrogen from the photosplitting of water.14 However, pure Fe3N exhibited relatively low photocatalytic activity under visible light. This low catalytic activity may be due to the large bulk morphology of the Fe3N NPs.
To improve the reactivity, transition metals can be applied to modify Fe3N catalysts. Previous research has investigated the doping of transition metals such as Mn, Cr and Co into iron nitride materials15–18 However, these reports focused on studying the structure and the magnetic properties of these doped materials. Few research efforts have been expended on application aspects. Moreover, the chemical synthetic methods mentioned in these studies, which included direct-nitridation, high-temperature and high-pressure methods, have many disadvantages such as high consumption of energy and high toxicity. Therefore it is necessary and urgent to find an easy synthesis method to carry out further studies of performance. In addition, it was previously found that doping transition metals can influence the morphology,19 which may be due to the changes of magnetic properties. Moreover, to the best of our knowledge, there have been few reports about the synthesis of (Fe1−xNix)3N NPs.16
In this paper, we report a straightforward sol–gel method to synthesize (Fe1−xNix)3N NPs. The structures, morphology and magnetic properties of Fe3N and (Fe1−xNix)3N NPs are carefully investigated. Moreover, the ultraviolet/visible-light photocatalytic water-splitting properties of Fe3N with different Ni doping concentrations are discussed. By comparing the photocatalytic water splitting properties of Fe3N with those of (Fe1−xNix)3N NPs, we discovery a potential application for (Fe1−xNix)3N NPs as a low-cost catalyst of photocatalytic water splitting.
Experiment section
Synthesis of the (Fe1−xNix)3N nanoparticles
Fe3N NPs doped with different concentrations of nickel were prepared in a horizontal tube furnace. Anhydrous ferric chloride (FeCl3), nickel chloride hexahydrate (NiCl2·6H2O), and ethanediamine were used as the source materials for the elements Fe, Ni and N, respectively, with acetone as a solvent. In detail, nickelous chloride hexahydrate and anhydrous ferric chloride were dissolved in 10 mL of acetone with vigorous stirring. Then 20 mL of ethylene diamine was added to the solution. The filtered precipitate was dried under a nitrogen atmosphere. After drying, black materials were obtained, and were used as precursors for subsequent reactions. These precursors were placed in the tube furnace, which was then heated to 580 °C and kept at this temperature for 30 min with a continuous nitrogen flow. The system was allowed to cool down to room temperature, and the resulting products were collected by grinding the mixture, cleaning the products with deionized water and ethanol several times, and then drying them in an oven overnight (60 °C) for further characterizations.
Characterizations
X-ray diffraction (XRD) patterns were recorded to analyze the phase and crystal structure of the as-synthesized samples using a Shimadzu XRD-6100, operated at a step size of 0.06° in the range of 30–70° and with Cu Kalpha radiation (λ = 0.15405 nm). Images of the nanostructured materials were obtained by using a Philips Tecnai G2 F20 high-resolution transmission electron microscope (HRTEM). X-ray photoelectron spectroscopy (XPS) was conducted on a Thermo VG Scientific Escalab 250 spectrometer with a monochromated Al Kalpha excitation source. A vibrating sample magnetometer (VSM, Lake Shore 7404) was used to characterize the magnetic properties of the as-synthesized samples. A magnetic hysteresis curve was obtained by varying the magnetic field between +17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Oe and −17000 Oe.
000 Oe and −17000 Oe.
Photocatalytic H2 production activity
Photocatalytic H2 production experiments were accomplished under Xe lamp irradiation at room temperature and chamber pressure. Reactions were carried out in a 100 mL flat-bottom flask with a quartz plate instead of its original glass, and the openings of the flask were sealed with a rubber plug. We used a 300 W Xe arc lamp (5 cm away from the reactor) with a UV-light source to carry out the photocatalytic reaction. The irradiated area was about 16.61 cm2. In order to maintain anaerobic conditions in the reaction system, suspensions of the photocatalyst were dispersed by using an ultrasonic bath and nitrogen was bubbled in for 30 min to completely remove the dissolved oxygen before irradiation. Typically, photocatalyst (40 mg) was dispersed under constant stirring in a mixed solution of triethanolamine (10 mL) and water (90 mL). The reaction solution was stirred continuously during the entire duration of the photocatalytic H2 production experiment, in order to keep the photocatalyst particles in a suspension. A volume of 0.2 mL of hydrogen gas was intermittently sampled through the rubber plug and was analyzed by using gas chromatography (GC-2014C TCD, Shimadzu, Japan, with nitrogen as a carrier gas and 5 Å molecular sieve column). All glassware was carefully washed with deionized water prior to use.
Results and discussion
Structural analysis
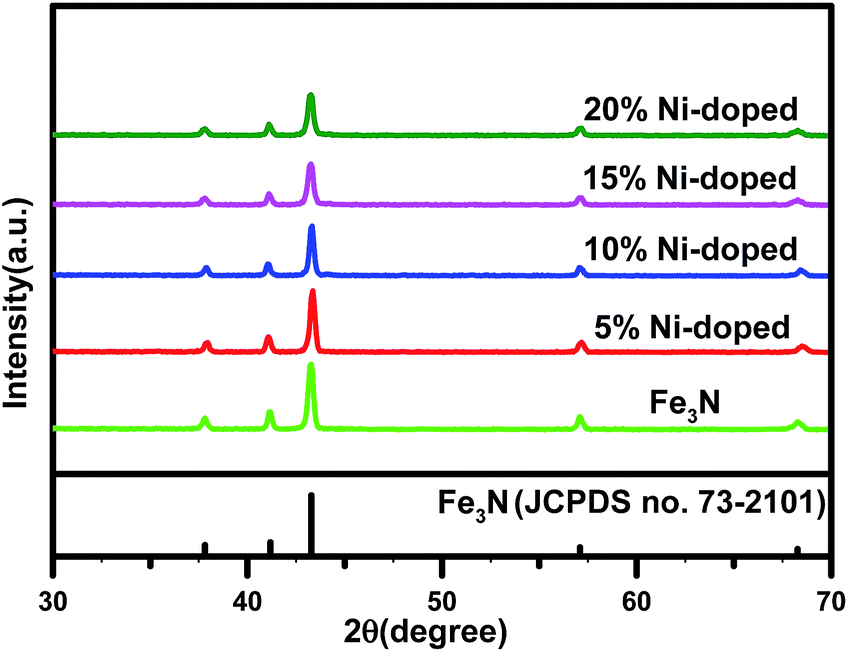
Fig. 1 shows the XRD patterns of the undoped Fe3N sample and the (Fe1−xNix)3N samples with Ni concentrations of 5%, 10%, 15% and 20%. The characteristic diffraction peaks at 2θ = 38.13°, 41.05°, 43.52°, 57.27° and 68.92° can be indexed to the (110), (002), (111), (112) and (300) crystal planes of ε-Fe3N, corresponding to JCPDS no. 73-2101. The XRD patterns of the undoped Fe3N and all of the doped Fe3N samples showed similar 2θ locations for their peaks, indicating that Ni substituted for Fe in the Fe3N lattice.
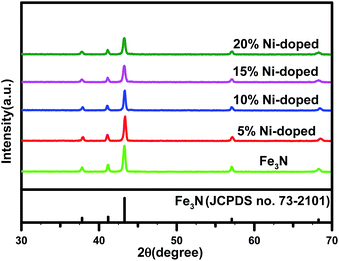 |
| | Fig. 1 XRD patterns of Fe3N and (Fe1−xNix)3N NPs. | |
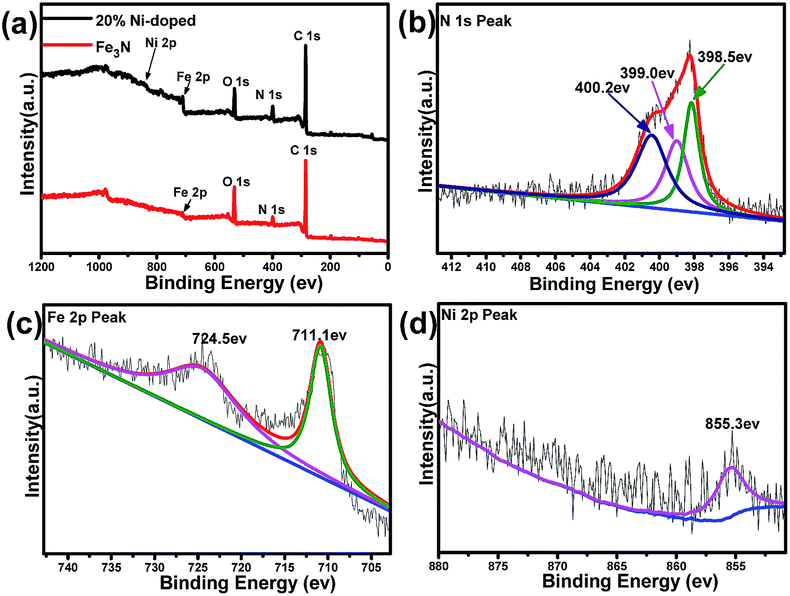
X-ray photoelectron spectroscopy (XPS) is conducted to identify the surface nature of the Fe3N and (Fe0.8Ni0.2)3N samples. As shown in Fig. 2a, the XPS spectrum of the Fe3N sample indicate it to be mainly composed of C, N, O and Fe elements, and no other impurity is observed, consistent with the results of XRD. In addition to the above elements, the spectrum of the (Fe0.8Ni0.2)3N show a Ni 2p peak, further proving that Fe is substituted with Ni in the Fe3N lattice. Fig. 2b shows the high-resolution N 1s spectrum of the 20% Ni-doped (Fe1−xNix)3N. According to this figure, the N elements could be divided into three types: the fitted peak of the N 1s spectrum located at 399.0 eV indicate that the N atoms combined with Fe atoms,20 confirming the formation of Fe3N; the peak at 398.5 eV could be ascribed to pyridinic-N; and another peak at 400.2 eV could be assigned to pyrrolic-N.21–23 These observations indicated that simultaneous N-doping from ethylenediamine carbonization in graphitic carbon occurred during the nitridation process. As shown in Fig. 2c, the centers of the electron binding energy peaks of Fe 2p3/2 and Fe 2p1/2 are found to be at 711.1 and 724.5 eV, respectively.24 As shown in the Ni 2p spectrum (in Fig. 2d), the binding energy peak at 855.3 eV is assigned to the Ni 2p3/2 peaks, confirming the presence of the Ni2+ ion and providing further evidence for the replacement of iron ions with Ni2+ ions in the crystal lattices of Fe3N.25–28
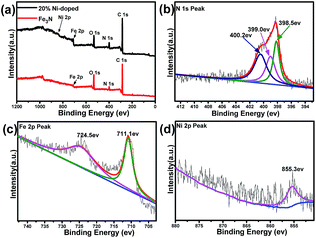 |
| | Fig. 2 Full XPS spectra of Fe3N and (Fe0.8Ni0.2)3N (a). High-resolution XPS spectra of the N 1s (b), Fe 2p (c) and Ni 2p (d) regions of the as-prepared (Fe0.8Ni0.2)3N. | |
Morphology
A transmission electron microscopy (TEM) image acquired for (Fe0.8Ni0.2)3N NPs (Fig. 3a) shows different shapes, such as ellipsoids, cubes, etc. But most of the nanoparticles are observed to be spherical, and quite dispersed in the carbon matrix, without any obvious agglomeration. Fig. 3b shows a typical high-magnification image of the sample. This image indicates that the nanoparticles are made up of a core–shell structure and the diameters of the as-prepared nanoparticles were not uniform. Fig. S2† shows the diameters of the nanoparticles to have varied from 5 to 50 nm with an average diameter of ∼18.7 nm. A typical high-magnification image of a single particle is shown in Fig. 3c, which reveal that the carbon layers with a well-ordered arrangement are close together and formed graphite shells surrounding the (Fe1−xNix)3N NPs. Well-crystallized structures with graphite lattice fringes of about 0.34 nm are observed, in accordance with the d002 value of graphite. Moreover, the thickness of the graphite shell is measured to be about 3.96 nm. Previous studies (but not discussed here) shows that these graphite shells were of importance for mass transport and hence for the electrocatalytic performance of the ORR and HER.29–31 Fig. 3d shows the nanoparticles with a carbon shell, and a well-defined crystalline lattice spacing of 0.21 nm in the core, corresponding to an interplanar spacing of the (111) diffraction peak of Fe3N. What is more, the TEM image of Fe3N NPs shown in Fig. S3† indicates the diameters of the Fe3N NPs to be much larger than that of the (Fe0.8Ni0.2)3N NPs, and no graphite shell is observed in this image. Moreover, the Fe3N NPs are observed to be irregularly shaped.
 |
| | Fig. 3 HRTEM images of (Fe0.8Ni0.2)3N (a–d). | |
Magnetic characterization
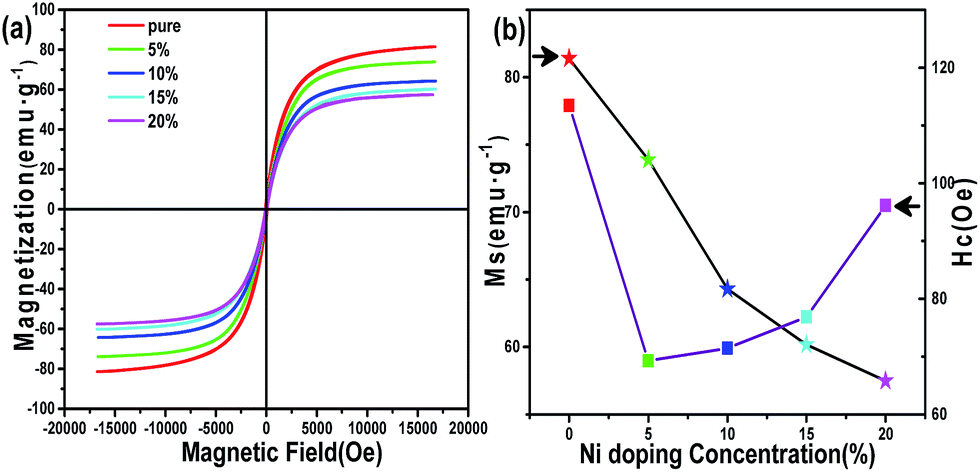
According to previous research, Fe3N NPs synthesized chemically rarely achieved high saturation magnetization (MS),32,33 mainly due to oxidation of the Fe3N surface. Fig. 4a shows the magnetization hysteresis loops for the Fe3N NPs and (Fe1−xNix)3N NPs of our synthetic method at room temperature. Both Fe3N and (Fe1−xNix)3N NPs show good soft magnetic behaviors. These excellent magnetic properties may be ascribed to the carbon shell capsulated on the surface of the core having protected the core from oxidation. However, all of the saturation magnetization values for both Fe3N and (Fe1−xNix)3N are found to be smaller than the theoretical ∼123 emu g−1 value of the bulk materials.34 This difference can also be ascribed to all of our samples having a massive carbon matrix capsulating the Fe3N and (Fe1−xNix)3N NPs. The MS values are clearly observed to decrease as the Ni doping concentration was increased, as shown in Fig. 4b. This relationship can be attributed to the ionic magnetic moment of Ni being less than that of Fe. Nevertheless, the values of HCi (coercivity) show an irregular trend with different doping concentrations.35 This trend can be attributed to the changes in the particle size caused by different doping concentrations.
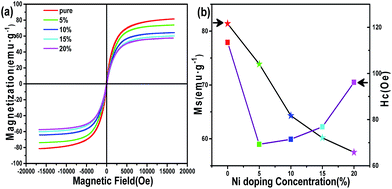 |
| | Fig. 4 (a) M–H plots of the Fe3N and (Fe1−xNix)3N NPs. (b) Graphs of the MS and HC vs. doping concentration. | |
Photocatalytic H2-production activity
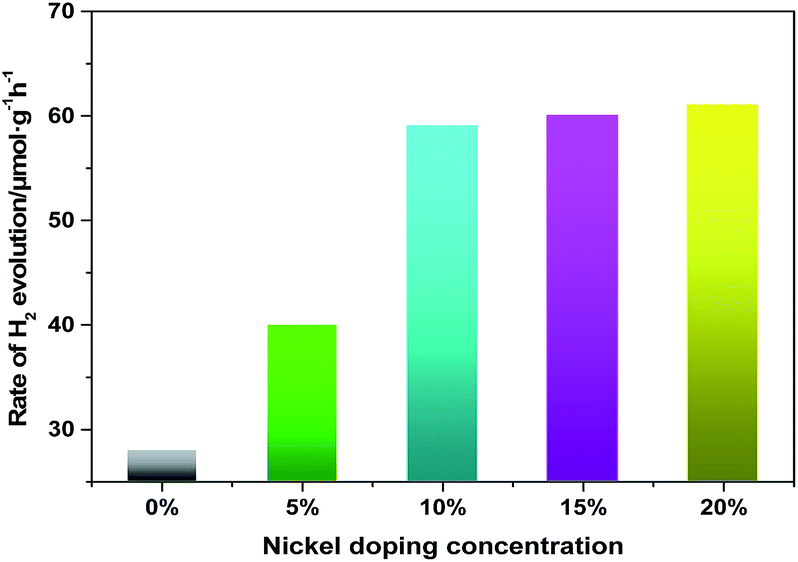
In our previous research, it has been reported that the pure Fe3N could be used as a photocatalyst under visible light. In the current work, we study the photocatalytic activity of pure Fe3N under ultraviolet and visible light. Moreover, we also find (Fe1−xNix)3N to have both ultraviolet- and visible-light photocatalytic activity. The photocatalytic activities of the samples are evaluated by measuring the H2 evolution under Xe lamp irradiation, using TEOA as a scavenger. Fig. 5 shows the ultraviolet- and visible-light photocatalytic activities of H2-production at various Ni doping concentrations. Pure Fe3N display relatively low photocatalytic ability under ultraviolet and visible light, at about 30.0 μmol g−1 h−1. However, when the doping concentration of Ni is 5%, the photocatalytic performance improve, to about 36.0 μmol g−1 h−1. In addition, we find that the photocatalytic activity improved to an effective level, at about 58.0 μmol g−1 h−1 when the doping concentration is increased to 10%. However, no further improvements in photocatalytic ability are observed with any further increases in the doping concentration. Therefore, a certain doping concentration of nickel is beneficial for photocatalytic H2 evolution. This effect of doped Ni could be explained in a couple of ways. (i) The doped nanoparticles are observed to have diameters much smaller than those of the pure sample, and would thus have many more active sites for the catalytic reaction. (ii) The lower saturation magnetization of the doped samples would decrease their agglomeration, hence improving their dispersion in water. In addition, in order to further verify the stability of the doped samples after the photocatalytic reaction, we analyze the XRD patterns of the doped samples (Fe0.9Ni0.1)3N and (Fe0.8Ni0.2)3N collected by centrifugation and drying. The test results (Fig. S4†) shows that there are no great changes in the catalytic process, besides the presence of Fe3O4 as a minor impurity phase.
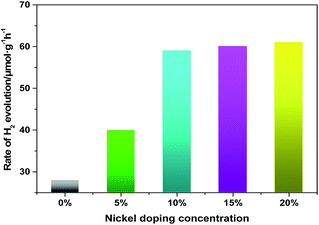 |
| | Fig. 5 Photocatalytic activities of Fe3N doped with 0%, 5%, 10%, 15% and 20% Ni for H2 production using triethanolamine as a scavenger under 500 W Xe lamp irradiation. | |
Conclusions
In the current work, we synthesized Fe3N NPs of different nickel doping concentration by a simple sol–gel route. XRD analysis of these nanoparticles show that there is no impurity in the doped samples, indicating that Fe of the Fe3N lattice is substituted by Ni. XPS analysis provide further evidence for the replacement of Ni2+ ions in the crystal lattices of Fe3N. It is shown that the (Fe0.8Ni0.2)3N sample adopt a spherical shape, and that these spheres are encapsulated in graphitic shells from the TEM investigation. Magnetic tests indicate that the Fe3N and (Fe1−xNix)3N NPs all show good soft magnetic behaviors. Moreover, the MS values tended to decrease as the Ni doping concentration is increased. We compare the photocatalytic water-splitting properties of Fe3N NPs and (Fe1−xNix)3N NPs and test their stability. The results suggest that (Fe1−xNix)3N NPs has potential application as a low-cost photocatalyst of the water-splitting reaction.
Acknowledgements
This work was supported by the National Natural Science Foundation of China.
Notes and references
- X. Chen, S. Shen, L. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503–6570 CrossRef CAS PubMed.
- Y. Bu, Z. Chen and C. Sun, Appl. Catal., B, 2015, 179, 363–371 CrossRef CAS.
- G. Li, Y. Wang and L. Mao, RSC Adv., 2014, 4, 53649–53661 RSC.
- Y. Wang, H.-B. Fang, Y.-Z. Zheng, R. Ye, X. Tao and J.-F. Chen, Nanoscale, 2015, 7, 19118–19128 RSC.
- S. Zhang, C. Chang, Z. Huang, Y. Ma, W. Gao, J. Li and Y. Qu, ACS Catal., 2015, 5, 6481–6488 CrossRef CAS.
- X. Yang, S. He, Y. Shu, Z. Shi, Y. Guo, Q. Gao and Y. Tang, RSC Adv., 2015, 5, 89282–89289 RSC.
- F. E. Osterloh, Chem. Soc. Rev., 2013, 42, 2294–2320 RSC.
- F. E. Osterloh, Chem. Mater., 2008, 20, 35–54 CrossRef CAS.
- Q. Gao, C. Giordano and M. Antonietti, Angew. Chem., Int. Ed., 2012, 51, 11740–11744 CrossRef CAS PubMed.
- W. Wang, P. Zhang, X. Wang, X. Lei, H. Ding and H. Yang, RSC Adv., 2015, 5, 90698–90704 RSC.
- P. Dhanasekaran, H. G. Salunke and N. M. Gupta, J. Phys. Chem. C, 2012, 116, 12156–12164 CAS.
- M. G. Kibria, F. A. Chowdhury, S. Zhao, B. AlOtaibi, M. L. Trudeau, H. Guo and Z. Mi, Nat. Commun., 2015, 6, 6797 CrossRef CAS PubMed.
- Q. Gao, N. Liu, S. Wang and Y. Tang, Nanoscale, 2014, 6, 14106–14120 RSC.
- P. Zhang, X. Wang, W. Wang, X. Lei, W. Yin and H. Yang, RSC Adv., 2015, 5, 68758–68764 RSC.
- N. S. Gajbhiye, R. S. Ningthoujam and S. Bhattacharyya, Hyperfine Interact., 2006, 164, 17–26 CrossRef CAS.
- K. Guo, D. Rau, L. Toffoletti, C. Mueller, U. Burkhardt, W. Schnelle, R. Niewa and U. Schwarz, Chem. Mater., 2012, 24, 4600–4606 CrossRef CAS.
- X. G. Ma, J. J. Jiang, P. Liang, J. Wang, Q. Ma and Q. K. Zhang, J. Alloys Compd., 2009, 480, 475–480 CrossRef CAS.
- S. El Khiraoui, M. Sajieddine, A. Vergnat, M. Sahlaoui, P. Bauer and A. Mabrouki, J. Alloys Compd., 2007, 440, 43–45 CrossRef CAS.
- X. Wang, P. Zhang, W. Wang, X. Lei and H. Yang, RSC Adv., 2015, 5, 57828–57832 RSC.
- Y. B. Li, Y. R. Yan, H. Ming and J. W. Zheng, Appl. Surf. Sci., 2014, 305, 683–688 CrossRef CAS.
- W. Yang, X. Liu, X. Yue, J. Jia and S. Guo, J. Am. Chem. Soc., 2015, 137, 1436–1439 CrossRef CAS PubMed.
- L. Wang, J. Yin, L. Zhao, C. Tian, P. Yu, J. Wang and H. Fu, Chem. Commun., 2013, 49, 3022–3024 RSC.
- H. Zhong, C. Deng, Y. Qiu, L. Yao and H. Zhang, J. Mater. Chem. A, 2014, 2, 17047–17057 CAS.
- G. Tong, Y. Liu, T. Wu, Y. Ye and C. Tong, Nanoscale, 2015, 7, 16493–16503 RSC.
- X. Long, G. Li, Z. Wang, H. Zhu, T. Zhang, S. Xiao, W. Guo and S. Yang, J. Am. Chem. Soc., 2015, 137, 11900–11903 CrossRef CAS PubMed.
- L. Bi, D. Xu, L. Zhang, Y. Lin, D. Wang and T. Xie, Phys. Chem. Chem. Phys., 2015, 17, 29899–29905 RSC.
- L. Huang, D. Chen, Y. Ding, Z. L. Wang, Z. Zeng and M. Liu, ACS Appl. Mater. Interfaces, 2013, 5, 11159–11162 CAS.
- C. Yuan, J. Li, L. Hou, X. Zhang, L. Shen and X. W. Lou, Adv. Funct. Mater., 2012, 22, 4592–4597 CrossRef CAS.
- X. Fan, Z. Peng, R. Ye, H. Zhou and X. Guo, ACS Nano, 2015, 9, 7407–7418 CrossRef CAS PubMed.
- Z.-Y. Wu, X.-X. Xu, B.-C. Hu, H.-W. Liang, Y. Lin, L.-F. Chen and S.-H. Yu, Angew. Chem., Int. Ed., 2015, 54, 8179–8183 CrossRef CAS PubMed.
- G. Zhong, H. Wang, H. Yu and F. Peng, J. Power Sources, 2015, 286, 495–503 CrossRef CAS.
- J. Theerthagiri, S. B. Dalavi, M. M. Raja and R. N. Panda, Mater. Res. Bull., 2013, 48, 4444–4448 CrossRef CAS.
- N. S. Gajbhiye, S. Bhattacharyya and S. M. Shivaprasad, Mater. Res. Bull., 2008, 43, 272–283 CrossRef CAS.
- M. Robbins and J. White, J. Phys. Chem. Solids, 1964, 25, 717–720 CrossRef CAS.
- S. Bhattacharyya, J. Phys. Chem. C, 2015, 119, 1601–1622 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra05990b |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.