
Single step synthesis of a polymer supported palladium composite: a potential anode catalyst for the application of methanol oxidation†
Abstract
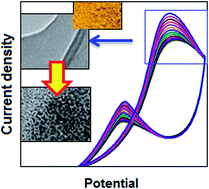
Polymer supported ionic palladium has been synthesized using a single step, in situ polymerization and composite formation route from the corresponding monomer and metal salt precursors. The composite has been characterized using various optical, microscopic and surface characterization techniques. The synthesized material was successfully used as the electrocatalyst for the methanol oxidation reaction in alkaline media, suggesting a potential candidate for methanol fuel cells application. During the reaction, the in situ transformation from ionic palladium to palladium nanoparticles has been noticed and this plays a major role for the significant improvement of the oxidation process.
 Please wait while we load your content...
Please wait while we load your content...

