
Tuning the composition of gold–silver bimetallic nanoparticles for the electrochemical reduction of hydrogen peroxide and nitrobenzene†
Abstract
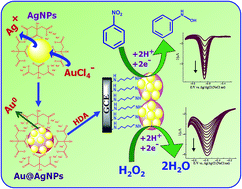
This paper reports the synthesis of gold–silver bimetallic nanoparticles (Au–AgNPs) with different Ag : Au compositions in an aqueous medium and their attachment on a glassy carbon electrode (GCE) via a 1,6-hexadiamine (HDA) linker for the electrochemical reduction of hydrogen peroxide (HP) and nitrobenzene (NB). Initially, silver nanoparticles (AgNPs) were synthesized by the reduction of silver nitrate using trisodium citrate as a capping agent and sodium borohydride as a reducing agent. Then, the Au–AgNPs were prepared by the galvanic displacement of Ag(0) by AuCl4− ions. The composition of the Au–AgNPs was varied by changing the mole ratio of Ag : Au in the range of 1 : 0 to 1 : 0.16. TEM images show that the Au–AgNPs were spherical in shape with a diameter of ∼16 nm. The prepared colloidal solution of Au–AgNPs were then attached on a HDA modified GCE through the Michael's addition reaction and were confirmed by UV-vis diffuse reflectance spectroscopy (DRS), atomic force microscopy (AFM), line scanning analysis, cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS). The AFM image shows that the Au–AgNPs were densely packed on the electrode surface. The Au–AgNPs modified electrode exhibits a higher heterogeneous electron transfer rate constant of 2.77 × 10−7 cm s−1 when compared to Ag and AuNPs modified electrodes. Furthermore, the electrocatalytic activity of the Au–AgNPs modified electrode was examined by studying the reduction of HP and NB. It was found that the Au–AgNPs with the Ag : Au mole ratio of 1 : 0.12 showed excellent electrocatalytic activity towards the reduction of both HP and NB by not only shifting their reduction potentials toward less negative potentials but also enhanced their currents compared to the bare GCE, Ag and AuNPs modified electrodes and Au–AgNPs of other molar ratios. The present modified electrode shows the limit of detection of 0.12 and 0.23 μM (S/N = 3) for HP and NB, respectively.
 Please wait while we load your content...
Please wait while we load your content...

