
A through bond energy transfer based ratiometric probe for fluorescent imaging of Sn2+ ions in living cells†
Susanta Adhikari*,
Avijit Ghosh,
Subhajit Guria and
Animesh Sahana
Department of Chemistry, University of Calcutta, 92, A.P.C. Road, Kolkata 700 009, West Bengal, India. E-mail: adhikarisusanta@yahoo.com; Fax: +91 33 23519755; Tel: +91 33 23509937
First published on 14th April 2016
Abstract
A 4-(naphthalen-1-ylethynyl) aniline appended rhodamine based fluorescent chemosensor ‘NAP-RD’ is synthesized which undergoes through bond energy transfer in the presence of Sn2+ ions in mixed aqueous media. NAP-RD is an excellent colorimetric and fluorescence ratiometric probe which recognizes traces of Sn2+ in real samples and a fluorescence indicator in living RAW264.7 cells. The new sensing probe NAP-RD shows higher sensitivity as well as faster response compared to the reported systems. The detection limit of NAP-RD is 5 × 10−9 M. DFT studies strongly support the experimental results.
1. Introduction
Ions of tin (Sn), a type of heavy metal, are usually found in the environment at low levels. Tin is an essential trace mineral for humans and is found in the greatest amount in the adrenal glands, liver, brain, spleen and thyroid gland.1,2 It is involved in several biochemical processes at the cellular level.3 Tin(II) as chloride, fluoride or citrate complexes is present in a number of consumables such as dental care products, soft drinks and food stuffs. SnCl2, a useful catalyst and reducing agent, is used for the synthesis of at least two bio-important moieties viz. indole and coumarin, in the preparation of many technetium-99m (99mTc) labelled radiopharmaceuticals in the field of nuclear medicine, fabrication of graphene/SnO2 (GS) nanocomposites and reduction of methyl ester during the preparation of PGF2α.4–10On the other hand, as an essential trace element, deficiency of tin can increase the risk factors associated with poor growth, hearing loss, and cancer prevention, but as a heavy metal, excess tin accumulation can detrimentally affect respiratory, nervous and digestive systems.11,12 Literature reports also revealed that, in forms of SnCl2, Sn2+ can be readily taken up by human white blood cells and cause DNA damage.13 SnCl2 is able to penetrate cells by calcium channels and this could be the mechanism responsible for DNA damage even in the absence of cellular commitment. Therefore, much attention has been given on the role of Sn2+ ion in biological systems and its cytotoxicity in recent years.14 So, because of its mixed blessing, it is of great importance to establish a method for the determination of Sn2+ ion in biological systems. Techniques such as flame atomic absorption spectrometry, potentiometric membrane sensing and UV/visible spectrophotometry have been developed for determination of Sn2+ ion. However, these methods are not suitable for in situ detection of Sn2+ in biological systems.15–17 In recent years, significant emphasis has been placed on the development of new, highly selective fluorescent chemosensors of different architectures for metal cations.18–23 Even though several fluorescent sensors have been developed for detection of biologically and environmentally important analytes, only a handful of intensity based sensors are known for Sn2+ ion.24–26 Therefore, there is essential need for development of probe which selectively detects Sn2+ ion with minimum background noise and high fluorescence intensity in cellular study.
In intensity-based probes, emission efficiency is highly dependent on the excitation intensity, probe concentration, background interferences and environment. These interferences can be eliminated by employing ratiometric fluorescent probes,27–44 which allow the measurement of changes of the intensity ratio at two emission bands induced by metal ions. FRET (fluorescence resonance energy transfer) probes can give signals in the presence and absence of analyte which minimizes these factors associated with intensity based probes for accurate and quantitative detection of analyte.35–44 Therefore, FRET sensing is highly desirable in optical therapy, cell physiology and sensing of analytes in bio-systems.45 Although the limitation of FRET probes lies in the fact that they requires good spectral overlap between acceptor absorbance and donor emission. Thus limits the wave length difference between the donor emission and acceptor absorbance.
The dyes based on TBET (through bond energy transfer), have the donor linked directly by an electronically conjugated bond to an acceptor, and energy transfer occurs through a conjugated bond without the need for spectral overlap. The energy transfer occurs extremely fast (in picosecond or even femtosecond range) and highly efficiently (approaching 100%), which is not as severely affected by surrounding medium molecules as in the case of FRET process. TBET appears to be more suitable than FRET for design of an efficient ratiometric probe for bio imaging of metal ions. Several cassettes based on TBET has been synthesized since the mechanism came into limelight for analyte sensing.46–61 Here in for the first time we report a TBET based Sn2+ ion sensor based on 4-(naphthalen-1-ylethynyl) aniline appended rhodamine. It is well known that spirocyclic rhodamine derivatives are useful for selective sensing of analyte via strong red emission, high quantum yield, cell permeability and efficient FRET acceptor.35–42,47–57 The probe NAP-RD displays a colorimetric change from colourless to pink and ratiometric fluorescence change from blue to orange red. The target compound was synthesized from 1-iodo naphthalene with considerable yields. Scheme 1 shows the synthesis of NAP-RD.
 | ||
| Scheme 1 Synthetic route for NAP-RD (a) CH2Cl2, POCl3 (b) 2-methyl-3-butyn-2-ol, [Pd(PPh3)4], CuI, Et3N, THF; (c) NaH, toluene; (d) [Pd(PPh3)4], CuI, Et3N, THF; (e) CH2Cl2, Et3N (f) acetic anhydride, acetic acid. | ||
All the intermediates and probe have been characterized by 1H NMR, 13C NMR and QTOF-MS spectra (Fig. S1–S15, ESI†).
2. Experimental
2.1. Materials and equipment
All metal salts were used as either their nitrate or their chloride salts. Other chemicals were of analytical reagent grade and used without further purification except when specified. 1H NMR spectra were recorded at 300 and 75 MHz for 13C NMR using solvent peak as internal reference at 25 °C. Thin-layer chromatography (TLC) was carried out on 0.25 mm thick precoated silica plates, and spots were visualized under UV light. Milli-Q Millipore 18.2 MΩ cm−1 water was used whenever required. A Shimadzu Multi Spec 2450 spectrophotometer was used for recording UV-vis spectra. FTIR spectra were recorded on a Shimadzu FTIR (model IR Prestige 21 CE) spectrophotometer. Mass spectra were recorded using a QTOF XEVO-G2 mass spectrometer in ESI positive mode. The steady state emission and excitation spectra were recorded with a Hitachi F-4500 spectrofluorometer. A Systronics digital pH meter (model 335) was used for pH measurement. Fluorescence microscope images were captured with an Olympus IX81 microscope and processed using Image Pro Plus v 7.0 Software.The synthetic protocols of these probes are very simple and inexpensive. Reaction of 1-iodo naphthalene with 2-methyl-3-butyn-2-ol in presence of Pd(PPh3)4 and CuI in anhydrous THF gives compound 2 in high yield. Compound 3 was obtained by reaction of compound 2 with sodium hydride in toluene. Reaction of 1-iodo aniline with compound 3 in presence of Pd(PPh3)4 and CuI in anhydrous THF gives precursor amine (compound 4) in high yield. Finally, reaction of acid chloride of rhodamine (1) with compound 4 in presence of triethylamine gives the desired probe NAP-RD in high yield (details in ESI†).
3. Results and discussion
3.1. Fluorescent response for Sn2+ ion
Absorbance and fluorescence experiments to examine the ‘turn-on’ response of the probe NAP-RD have been performed in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 acetonitrile:HEPES buffer (10 mM, pH 7.4). The emission maximum of free NAP-RD is 442 nm (λex = 366 nm) which is attributed to the emission of the conjugated naphthyl moiety 4-(naphthalen-1-ylethynyl) aniline. In presence of Sn2+, it shows emission maxima at 599 nm (λex = 366 nm). The ratiometric nature of the curve is shown in Fig. 1. Upon gradual addition of Sn2+ ion the band at 442 nm continuously decreases and a new band at 599 nm increases significantly in due course. A plot of emission intensity vs. increasing Sn2+ ion has been shown in Fig. 1 inset.
:4 acetonitrile:HEPES buffer (10 mM, pH 7.4). The emission maximum of free NAP-RD is 442 nm (λex = 366 nm) which is attributed to the emission of the conjugated naphthyl moiety 4-(naphthalen-1-ylethynyl) aniline. In presence of Sn2+, it shows emission maxima at 599 nm (λex = 366 nm). The ratiometric nature of the curve is shown in Fig. 1. Upon gradual addition of Sn2+ ion the band at 442 nm continuously decreases and a new band at 599 nm increases significantly in due course. A plot of emission intensity vs. increasing Sn2+ ion has been shown in Fig. 1 inset.
 | ||
| Fig. 1 Changes in the emission spectra of NAP-RD (10 μM), upon gradual addition of Sn2+ (0.0, 0.005, 0.01, 0.05, 0.1, 0.5, 1.0, 5.0, 10.0, 20.0, 50.0, 75.0, 100.0, 200.0, 300.0 and 500.0 μM) in 1:4 (v/v) acetonitrile–HEPES (10 μM, pH = 7.4; λex = 366 nm, inset: plot of emission intensity vs. concentration). | ||
Similarly, in absence of Sn2+ ion, one absorption band could be observed at 323 nm, which should be ascribed to the absorption profile of conjugated naphthyl moiety. However, the absorption band was red-shifted from 323 nm to 556 nm upon the addition of increasing amount of Sn2+, and the colour of the solution changed from colourless to pink, suggesting the ring-open form of NAP-RD (Fig. 2). Naked eye and UV-light exposed colour of Sn2+ adducts of NAP-RD are presented in Fig. S16 (ESI†). A plot of emission intensity vs. Sn2+ concentration, the linear region (up to 0.05 μM Sn2+, Fig. S17, ESI†) which is useful for determination of unknown Sn2+ concentration at low levels. The effect of pH on the emission intensities of free NAP-RD and Sn2+ bound systems (Fig. S18, ESI†) indicates the optimum pH ranging from 4.0 to 8.0 for Sn2+ detection. From Job's plot (Fig. S19, ESI†), 1:1 stoichiometry is confirmed. From this information, binding constant of NAP-RD with Sn2+ ion was calculated using Benesi–Hildebrand equation,62 (Fmax − F0)/(Fx − F0) = 1 + (1/K)(1/[M]n), where Fmax, F0, and Fx are fluorescence intensities of NAP-RD in the presence of Sn2+ at saturation, free NAP-RD and any intermediate Sn2+ concentration, (Fig. S20 in ESI†). A plot of (Fmax − F0)/(Fx − F0) vs. 1/[M]1 (here n = 1) yields the apparent binding constant value as 3.1 × 105 M−1 (R2 = 0.981).
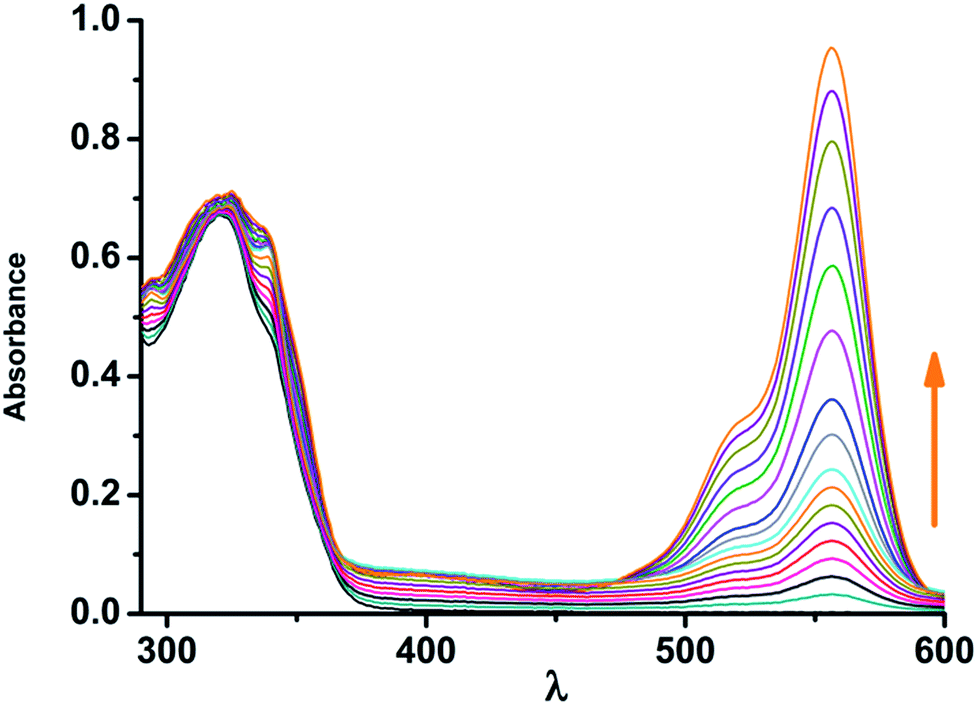 | ||
| Fig. 2 Absorbance of NAP-RD (10 μM) toward increasing amount of Sn2+ (0–200 μM) in 1:4 acetonitrile:HEPES buffer (10 mM, pH 7.4) λex = 366 nm. | ||
The detection limit was ascribed to be 5 × 10−9 M (5 nM) of Sn2+ for NAP-RD. The quantum yield of the probe NAP-RD and its Sn2+ adduct were 0.02 and 0.330. The new sensing probe NAP-RD shows higher sensitivity as well as faster response compared to the reported systems.24–26 A table of comparison of the detection limits with the reported probes has been shown in Table S1.† The emission of compound 4 (Fig. S21, in ESI†) at ∼512 nm being close to the absorbance of open ring rhodamine B moiety (∼550 nm), significant overlap and, hence efficient FRET process is anticipated (Fig. S22, in ESI†). On the other hand, the emission intensity of compound 5 situated at 420 nm (Fig. S23, in ESI†), is not at all close to the absorbance of the ring opened form of rhodamine B moiety and hence no significant overlap is predicted. TBET is not a subject to the constraint of spectral overlap between the donor emission and the acceptor absorption.57 It may, therefore, be possible to separate the donor absorption from the acceptor fluorescence by many wave numbers. In TBET systems the donor and acceptor are joined by a conjugated spacer, which prevents them from becoming flat and conjugated.46–61 These types of systems absorb at a wavelength characteristic of a donor then emit via a receptor.
In NAP-RD, 4-(naphthalen-1-ylethynyl) aniline derivative (served as the energy donor) and ring opened form of rhodamine B (served as the energy acceptor) were covalently linked through an amide bond (Fig. 3). NAP-RD shows a blue fluorescence emission resulting from the 4-(naphthalen-1-ylethynyl) aniline moiety, and no fluorescence emission produced from the rhodamine B moiety due to the ring-closing of rhodamine B. Thus no TBET was observed. Upon addition of Sn2+ ion, the spirolactam ring of rhodamine unit opened and consequently transferred energy from 4-(naphthalen-1-ylethynyl) aniline to rhodamine unit. A strong fluorescence emission is observed being associated with the conjugated system.
 | ||
| Fig. 3 Proposed mechanism of Sn2+ mediated spirolactam ring opening in NAP-RD and through bond energy transfer of the resulting cassette. | ||
3.2. Energy transfer efficiency (ETE) calculations
To estimate the energy transfer efficiency (ETE) of NAP-RD upon addition of Sn2+, a model compound (donor), 5 was synthesized (Scheme 1). Compound 5 showed a strong fluorescence emission centralized at 420 nm and there were no changes in the fluorescence intensity upon addition of Sn2+ compound. These results indicate that Sn2+ has no effect on the fluorescence emission of compound 5, and therefore an effective TBET process between the donor and acceptor occurred. The ETE of NAP-RD for Sn2+ was calculated to be 94.22% (Fig. 1, S23 and S25, ESI†). Many pioneering reports on TBET system indicate that there is always some FRET process accompanied with TBET, resulting in the failure of 100% energy transfer efficiency.42–57Furthermore, for practical applications, it is very important that the fluorescence intensity of the acceptor in the cassette is greater than that of the acceptor without a pendent donor when it is excited at the donor absorption wavelength. A high selectivity towards target metal ion over other metal cations and anions is also necessary for a probe with potential application in complex biological or environmental systems. Upon addition of 0.05 μM of Sn2+ into NAP-RD (Fig. 4), induces a significant enhancement of fluorescence ratio at I599/I442. Other common metal cations or anions at 5 μM did not induce obvious changes of fluorescence ratio. Fe3+ in a concentration higher than 20 μM showed a slight interference. All these selective results indicate that the proposed probe could meet the selective requirements for biomedical and environmental applications.
 | ||
| Fig. 4 Fluorescence response of NAP-RD (10 μM) to 0.5 μM of Sn2+ or 5.0 μM of other metal ions (the black bar portion) and to the mixture of 5.0 μM of other metal ions with 0.5 μM of Sn2+ (the red bar portion). Metal ions: Sn2+ (1), Hg2+ (2), Zn2+ (3), Pb2+ (4), Mn2+ (5), Ag+ (6), Au+ (7), Co2+ (8), Al3+ (9), Ni2+ (10), Ca2+ (11), Cu2+ (12), Cd2+ (13), Cr3+ (14) and Fe3+ (15). | ||
3.3. Binding mode NAP-RD of with Sn2+
In order to support the above mechanism for the TBET based probe, 1H NMR titration has been performed by adding Sn2+ to the MeOH-d4 solution of NAP-RD (Fig. 5). | ||
| Fig. 5 (I) 1H NMR spectrum of NAP-RD in MeOH-d4 (II) upon addition of 0.5 equivalent of Sn2+ ion in D2O and (III) upon addition of 1 equivalent of Sn2+ ion in D2O. | ||
On the other hand, significant spectral change of NAP-RD has been observed upon addition of Sn2+ from the 1H NMR titration experiment. Fig. 5(I) shows the 1H NMR of free NAP-RD. Upon addition of 0.5 equivalent of Sn2+ ion in D2O (II) the aromatic protons of the rhodamine (viz. p, q, r and l, m, n, o) and 4-(naphthalen-1-ylethynyl) aniline (viz. h, i, j, k and a–g) showed significant shifts. The p, q, r protons are downfield shifted from 6.31 ppm to 6.61 ppm. Proton ‘l’ downfield shifted from 7.86 to 7.94 ppm. The proton b, c also downfield shifted from 7.54 to 7.66 ppm. As expected, the ‘x’ protons corresponding to one of the two N,N-diethyl amine of rhodamine unit also downfield shifted significantly (3.23 ppm to 3.43 ppm). Therefore, Sn2+ mediated spirolactam ring opening of rhodamine unit was confirmed from the 1H NMR titration experiment also.
The QTOF-MS spectra of the resulting complex of [NAP-RD–Sn2+] were found to be 877.18567 (Fig. S26 in ESI†) attributed to the [NAP-RD–SnCl2 + H2O]. In Fig. 3 the possible binding mode of SnCl2 with NAP-RD are represented. The expanded elemental composition data from QTOF-MS spectra peak at 877.18567 (Fig. S27 in ESI†) also confirmed the splitting pattern of SnCl2 bound NAP-RD.
DFT studies further support Sn2+ assisted ‘Turn On’ response in NAP-RD. Density functional theory calculations of the energy gapes between HOMO (the highest occupied molecular orbitals) and LUMO (the lowest unoccupied molecular orbitals) of NAP-RD and Sn2+ bound complexes were performed. The observed energy gaps between HOMO and LUMO of Sn2+ complex of free NAP-RD and NAP-RD were calculated to be 2.26 eV and 3.62 eV, respectively. The energy optimized geometries with HOMO–LUMO of the probes (Table S2, ESI†) and Sn2+ adducts (Table S3, ESI†) have been generated using B3LYP/6-31G and B3LYP/LanL2DZ basis sets respectively with Gaussian 09 software.63 The HOMO–LUMO energy difference of [NAP-RD–Sn2+] complex is lower than that of free NAP-RD. The individual energy levels of HOMO and LUMO of the adduct complex are also low in energy.
The excited state related calculations of [NAP-RD–Sn2+] adduct has been carried out using Time-Dependent Density Function Theory (TDDFT), based on the optimized ground state geometry. From the calculated data, it is found that in [NAP-RD–Sn2+] two main electronic transition are observed mainly at 555 nm, 2.23395 eV, f = 0.7870 and 418 nm, 2.95905 eV and f = 0.387. The observed electronic transition at 555 nm is close to the experimental absorbance of [NAP-RD–Sn2+] adduct. The energy optimized geometry and the possible binding modes are summarized in Table S4.† The calculated coefficient of the wave function (CI) for the HOMO to LUMO transition is 0.8342 which indicates that radiative decay is possible and eventually leads to emission in each adduct (Fig. 6).
 | ||
| Fig. 6 Selected MOs and HOMO–LUMO energy difference NAP-RD and [NAP-RD + Sn2+]. | ||
Intracellular Sn2+ imaging using NAP-RD on RAW264.7 cells have been investigated. The cells are incubated with 10 μM NAP-RD for 2 h. Then the cells are washed thrice with PBS buffer and Sn2+ is added to the medium followed by further incubation for 1 h. Fig. 7 reveals that NAP-RD is cell permeable to efficiently image intracellular Sn2+ under fluorescence microscope (Fig. S28, ESI†).
 | ||
| Fig. 7 Fluorescence imaging of Sn2+ in RAW264.7 cells: (a) cells after incubation with 10 μM NAP-RD; (b) fluorescence image of those cells after incubation with 20 μM of Sn2+; (c) bright field images of those cells in panel b; (d) overlay image of b and c. Cell imaging experiment was performed under green light (480–550 nm) excitation with fluorescence signals collecting in the range of >590 nm. NAP-RD was prepared in ∼0.3% DMSO in water. | ||
The cytotoxicity of NAP-RD on RAW264 cells is determined by MTT assay (ESI†). Upon exposure of 10 μM NAP-RD for 12 h, ∼90% of the RAW264 cells remains viable. This nullifies the possibility of any significant cytotoxic influence of NAP-RD on RAW264 cells. Therefore, NAP-RD may be used as an ideal chemosensor for Sn2+ in living systems.
4. Conclusion
In summary, we have designed and synthesized through bond energy transfer based excellent colorimetric and fluorescent probe for selective detection of Sn2+ ion in aqueous medium. The new sensing probe NAP-RD shows higher sensitivity as well as faster response compared to the reported systems. The probe NAP-RD shows about 95% energy transfer efficiency. NAP-RD can effectively images RAW264.7 cell line. So, the present demonstration was a new addendum for important metal ion sensing in the horizon of rhodamine based receptors for cation recognition.Acknowledgements
A. G. is thankful to UGC and S. G. to CSIR, New Delhi for providing fellowship. S. A. acknowledges the SERB-DST (project no. SR/S1/OC-101/2012) for financial support.Notes and references
- H. Rudel, Ecotoxicol. Environ. Saf., 2003, 56, 180–189 CrossRef CAS PubMed.
- S. G. Schäfer and U. Femfert, Regul. Toxicol. Pharmacol., 1984, 4, 57–69 CrossRef.
- N. J. Snoeij, A. H. Penninks and W. Seinen, Environ. Res., 1987, 44, 335–353 CrossRef CAS PubMed.
- K. K. Upadhyay, R. K. Mishra and A. Kumar, Catal. Lett., 2008, 121, 118–120 CrossRef CAS.
- C. S. Cho, H. K. Lim, S. C. Shim, T. J. Kim and H. J. Choi, Chem. Commun., 1998, 995–996 RSC.
- J. Mattos, V. Matos, M. Rodrigues, M. Oliveira, F. Dantas, S. Filho, M. Filho and A. Araujo, Molecules, 2012, 17, 12974–12983 CrossRef PubMed.
- F. Li, J. Song, H. Yang, S. Gan, Q. Zhang, D. Han, A. Ivaska and L. Niu, Nanotechnology, 2009, 20, 455602–455607 CrossRef PubMed.
- M. Hamberg and B. Samuelsson, Proc. Natl. Acad. Sci. U. S. A., 1973, 70, 899–903 CrossRef CAS.
- S. Blunden and T. Wallace, Food Chem. Toxicol., 2003, 41, 1651–1662 CrossRef CAS PubMed.
- M. L. B. Assisa, J. B. C. Netob, J. E. Q. Souzac, A. Caldeira-de-Araújoa and M. B. Filho, Cancer Lett., 1998, 130, 127–131 CrossRef.
- K. A. Winship, Adverse Drug React. Acute Poisoning Rev., 1988, 7, 19–38 CAS.
- N. Cardarelli, Thymus, 1990, 15, 223–231 CAS.
- L. R. Sherman, J. Masters, R. Peterson and S. Levine, J. Anal. Toxicol., 1986, 10, 6–9 CrossRef CAS PubMed.
- C. M. Viau, T. N. Guecheva, F. G. Sousa, C. Pungartnik, M. Brendel, J. Saffi and J. A. Pegas Henriques, Arch. Toxicol., 2009, 83, 769–775 CrossRef CAS PubMed.
- M. Arvand, A. M. Moghimi, A. Afshari and N. Mahmoodi, Anal. Chim. Acta, 2006, 579, 102–108 CrossRef CAS PubMed.
- S. Ulusoy, H. I. Ulusoy and M. Akcay, Food Chem., 2012, 134, 419–426 CrossRef CAS.
- A. S. Dadda, A. C. Teixeira, P. K. Feltes, M. M. Campos, C. E. Leite and C. M. Moriguchi-Jeckel, J. Braz. Chem. Soc., 2014, 25, 1621–1629 CAS.
- Y. Yang, Q. Zhao, W. Feng and F. Li, Chem. Rev., 2013, 113, 192–270 CrossRef CAS PubMed.
- S. Adhikari, A. Ghosh, S. Mandal, A. Sahana and D. Das, RSC Adv., 2015, 5, 33878–33884 RSC.
- Y. Zhao, X. B. Zhang, Z. X. Han, L. Qiao, C. Y. Li, L. X. Jian, G. L. Shen and R. Q. Yu, Anal. Chem., 2009, 81, 7022–7030 CrossRef CAS PubMed.
- X. H. Zhao, R. M. Kong, X. B. Zhang, H. M. Meng, W. N. Liu, W. Tan, G. L. Shen and R. Q. Yu, Anal. Chem., 2011, 83, 5062–5066 CrossRef CAS PubMed.
- Z. Y. Ming, S. B. Bing, Z. Peng, H. J. Qiang, C. Pei, L. Qi, L. Jun and W. T. Bao, Sci. China: Chem., 2013, 56, 612–618 CrossRef.
- S. F. Ling, N. H. Fu, S. H. Ying, W. J. Yun and P. X. Jun, Sci. China: Chem., 2014, 57, 1043–1047 CrossRef.
- S. Adhikari, S. Mandal, A. Ghosh, S. Guria and D. Das, Dalton Trans., 2015, 44, 14388–14393 RSC.
- X. Bao, X. Cao, X. Nie, Y. Jin and B. Zhou, Molecules, 2014, 19, 7817–7831 CrossRef CAS PubMed.
- H. Lan, Y. Wen, Y. Shi, K. Liu, Y. Mao and T. Yi, Analyst, 2014, 139, 5223–5229 RSC.
- M. Baruah, W. Qin, R. A. L. Vallee, D. Beljonne, T. Rohand, W. Dehaen and N. Boens, Org. Lett., 2005, 7, 4377 CrossRef CAS PubMed.
- Z. C. Xu, Y. Xiao, X. H. Qian, J. N. Cui and D. W. Cui, Org. Lett., 2005, 7, 889–892 CrossRef CAS PubMed.
- C. Lu, Z. Xu, J. Cui, R. Zhang and X. Qian, J. Org. Chem., 2007, 72, 3554–3557 CrossRef CAS PubMed.
- Z. Xu, K. H. Baek, H. N. Kim, J. Cui, X. Qian, D. R. Spring, I. Shin and J. Yoon, J. Am. Chem. Soc., 2010, 132, 601–610 CrossRef CAS PubMed.
- X. Cheng, Q. Li, J. Qin and Z. Li, ACS Appl. Mater. Interfaces, 2010, 2, 1066–1072 CAS.
- B. C. Zhu, C. C. Gao, Y. Z. Zhao, C. Y. Liu, Y. M. Li, Q. Wei, Z. M. Ma, B. Du and X. L. Zhang, Chem. Commun., 2011, 47, 8656–8658 RSC.
- C. C. Woodroofe and S. J. Lippard, J. Am. Chem. Soc., 2003, 125, 11458–11459 CrossRef CAS PubMed.
- C. Y. Li, X. B. Zhang, L. Qiao, Y. Zhao, C. M. He, S. Y. Huan, L. M. Lu, L. X. Jian, G. L. Shen and R. Q. Yu, Anal. Chem., 2009, 81, 9993–10001 CrossRef CAS PubMed.
- M. Yuan, W. Zhou, X. Liu, M. Zhu, J. Li, X. Yin, H. Zheng, Z. Zuo, C. Ouyang, H. Liu, Y. Li and D. Zhu, J. Org. Chem., 2008, 73, 5008–5014 CrossRef CAS PubMed.
- M. H. Lee, H. J. Kim, S. W. Yoon, N. Park and J. S. Kim, Org. Lett., 2008, 10, 213–216 CrossRef CAS PubMed.
- Z. Zhou, M. Yu, H. Yang, K. W. Huang, F. Li, T. Yi and C. Huang, Chem. Commun., 2008, 3387–3389 RSC.
- X. Zhang, Y. Xiao and X. Qian, Angew. Chem., Int. Ed., 2008, 47, 8025–8029 CrossRef CAS PubMed.
- S. Adhikari, S. Mandal, A. Ghosh, P. Das and D. Das, J. Org. Chem., 2015, 80, 8530–8538 CrossRef CAS PubMed.
- Z. X. Han, X. B. Zhang, Z. Li, Y. J. Gong, X. Y. Wu, Z. Jin, C. M. He, L. X. Jian, J. Zhang, G. L. Shen and R. Q. Yu, Anal. Chem., 2010, 82, 3108–3113 CrossRef CAS PubMed.
- H. B. Yu, Y. Xiao, H. Y. Guo and X. H. Qian, Chem.–Eur. J., 2011, 17, 3179 CrossRef CAS PubMed.
- L. Yuan, W. Lin, K. Zheng and S. Zhu, Acc. Chem. Res., 2013, 46, 1462–1473 CrossRef CAS PubMed.
- V. S. Jisha, A. J. Thomas and D. Ramaiah, J. Org. Chem., 2009, 74, 6667–6673 CrossRef CAS PubMed.
- A. E. Albers, V. S. Okreglak and J. C. Chang, J. Am. Chem. Soc., 2006, 128, 9640–9641 CrossRef CAS PubMed.
- D. Geißler, S. Stufler, H. G. Löhmannsröben and N. Hildebrandt, J. Am. Chem. Soc., 2013, 135, 1102–1109 CrossRef PubMed.
- X. Zhang, Y. Xiao, L. He and Y. Zhang, J. Org. Chem., 2014, 79, 6315–6320 CrossRef CAS PubMed.
- J. Fan, M. Hu, P. Zhan and X. Peng, Chem. Soc. Rev., 2013, 42, 29–43 RSC.
- J. Fan, P. Zhan, M. Hu, W. Sun, J. Tang, J. Wang, S. Sun, F. Song and X. Peng, Org. Lett., 2013, 15, 492–495 CrossRef CAS PubMed.
- L. Zhou, X. Zhang, Q. Wang, Y. Lv, G. Mao, A. Luo, Y. Wu, Y. Wu, J. Zhang and W. Tan, J. Am. Chem. Soc., 2014, 136, 9838–9841 CrossRef CAS PubMed.
- H. W. Liu, X. B. Zhang, J. Zhang, Q. Q. Wang, X. X. Hu, P. Wang and W. Tan, Anal. Chem., 2015, 87, 8896–8903 CrossRef CAS PubMed.
- L. Zhou, Q. Wang, X. B. Zhang and W. Tan, Anal. Chem., 2015, 87, 4503–4507 CrossRef CAS PubMed.
- Y. J. Gong, X. B. Zhang, C. C. Zhang, A. L. Luo, T. Fu, W. Tan, G. L. Shen and R. Q. Yu, Anal. Chem., 2012, 84, 10777–10784 CrossRef CAS PubMed.
- S. Saha, P. Mahato, M. Baidya, S. K. Ghosh and A. Das, Chem. Commun., 2012, 48, 9293–9295 RSC.
- V. Bhalla, V. Vij, R. Tejpal, G. Singh and M. Kumar, Dalton Trans., 2013, 42, 4456–4463 RSC.
- G. U. Reddy, V. Ramu, S. Roy, N. Taye, S. Chattopadhyay and A. Das, Chem. Commun., 2014, 50, 14421–14424 RSC.
- V. Bhalla, Roopa, M. Kumar, P. R. Sharma and T. Kaur, Inorg. Chem., 2012, 51, 2150–2156 CrossRef CAS PubMed.
- W. Lin, L. Yuan, Z. Cao, Y. Feng and J. Song, Angew. Chem., Int. Ed., 2010, 49, 375–379 CrossRef CAS PubMed.
- D. Sain, C. Kumari, A. Kumar and S. Dey, Sens. Actuators, B, 2015, 221, 849–856 CrossRef CAS.
- X. Qu, Q. Liu, X. Ji, H. Chen, Z. Zhou and Z. Shen, Chem. Commun., 2012, 48, 4600–4602 RSC.
- G. Jiao, L. H. Thoresen and K. Burgess, J. Am. Chem. Soc., 2003, 125, 14668–14669 CrossRef CAS PubMed.
- R. Bandichhor, A. D. Petrescu, A. Vespa, A. B. Kier, F. Schroeder and K. Burgess, J. Am. Chem. Soc., 2006, 128, 10688–10689 CrossRef CAS PubMed.
- H. A. Benesi and J. H. Hildebrand, J. Am. Chem. Soc., 1949, 71, 2703–2707 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb and J. R. Cheeseman, et al., Gaussian 09, revision A.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H, 13C, ESI-MS, UV-Vis absorption spectra, cell imaging, DFT studies and additional spectroscopic data. See DOI: 10.1039/c6ra05650d |
| This journal is © The Royal Society of Chemistry 2016 |