
Chitosan-coated liposomes encapsulating curcumin: study of lipid–polysaccharide interactions and nanovesicle behavior
Abstract
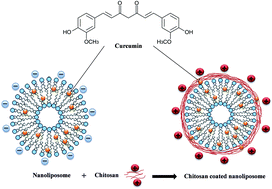
Despite various spectacular therapeutic properties, curcumin has low bioavailability mainly due to its poor solubility in water. In this paper, we encapsulated curcumin by nanoliposomes prepared from salmon purified phospholipid and coated with chitosan. Various techniques were used in order to study the interactions among phospholipid, chitosan and curcumin. FTIR results showed both electrostatic and hydrophobic interactions as well as hydrogen bonding between chitosan and phospholipid, while hydrophobic forces and hydrogen bonding dominated the interactions between curcumin and phospholipid as well as between curcumin and chitosan. Shear viscosity measurements demonstrated a flow behavior change from Newtonian to shear thinning after liposome coating. The increase/decrease stress ramp showed that the addition of chitosan layer decreased significantly the hysteresis loop area (thixotropic behavior) and therefore increased significantly the liposomal dispersion stability. The viscoelastic properties investigated by small-amplitude oscillatory shear rheology demonstrated improvement of mechanical stability after chitosan addition. Small-angle X-ray scattering experiments revealed that the liposome membrane structure was not affected by the chitosan layer or the encapsulated curcumin.
 Please wait while we load your content...
Please wait while we load your content...

