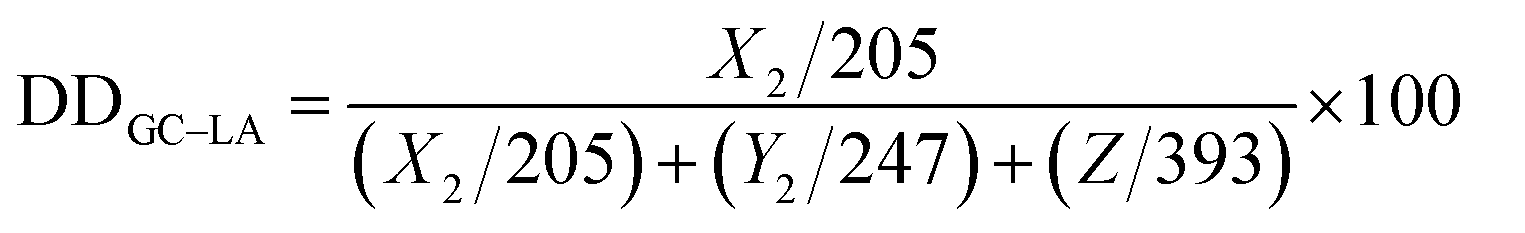DOI:
10.1039/C6RA05501J
(Paper)
RSC Adv., 2016,
6, 31391-31400
Reduction-responsive core-crosslinked micelles based on a glycol chitosan–lipoic acid conjugate for triggered release of doxorubicin†
Received
1st March 2016
, Accepted 21st March 2016
First published on 22nd March 2016
Abstract
Reduction-responsive core-crosslinked micelles were developed based on a glycol chitosan–lipoic acid (GC–LA) conjugate and used for triggered release of doxorubicin (DOX). The substitution degree of GC–LA was 8.3 lipoic acid groups per 100 sugar units of glycol chitosan. GC–LA could form nanoscaled micelles in aqueous solution, wherein the critical micelle concentration (CMC) of 0.081 mg mL−1 was determined. Furthermore GC–LA micelles can be crosslinked by a catalytic amount of dithiothreitol. The mean diameter of DOX-loaded core-crosslinked GC–LA (DOX-GC–LA/cc) micelles increased from 305 to 408 nm as the DOX-loading content increased from 6.03% to 10.74%. DOX-loaded crosslinked micelles demonstrated obvious reduction-triggered destabilization. DOX release from non-crosslinked GC–LA micelles was 87.6% for up to 96 h, whereas 25.3% of DOX release from DOX-GC–LA/cc micelles was observed in phosphate buffered saline (PBS, pH 7.4). Notably, in the presence of a 20 mM GSH-containing environment, accelerated DOX release from DOX-GC–LA/cc micelles was found. The blank micelles had low cytotoxicity in vitro, and DOX-GC–LA/cc micelles demonstrated intracellular redox-responsive characteristics in A549 cancer cells. These results suggested that GC–LA core-crosslinked micelles could be promising carriers for anticancer drug delivery.
1 Introduction
In the past decade, a tremendous effort has been concentrated on the development of polymer nanoparticles for the controlled delivery of anticancer drugs.1–6 Polymeric micelles can self-assemble into supramolecular aggregates and form a typical core–shell architecture in an aqueous medium.7,8 These nanocarriers exhibit unique features, such as passive targeting in tumors via the enhanced permeability and retention (EPR) effect, improving drug bioavailability and minimizing side effect.9
However, it should be noted that polymeric micelles in vivo may be unstable because of the large dilution volume and/or interactions with cells and biomolecules present in the blood, resulting in destructive core–shell architecture and premature drug release. In order to improve the structural integrity and stability of micelles, a promising approach is to covalently crosslink polymer micelles, such as shell or core crosslinking.10–14 Among them, an appealing crosslinking way is the use of disulfide linkage, which demonstrates excellent biocompatibility and reduction sensitivity. Disulfide linkage is stable in the process of blood circulation and the extracellular environment but destroyed under the reducing environment, especially within tumor cells. An endogenous reducing agent, glutathione (GSH), effectively reacted with thiol–disulfide bonds. GSH is presented at much higher level in the cytosol of tissue cells (1–10 mM) than the extracellular environment (2–10 μM).15,16 What's more, its level in the tumor cells is 7–10 fold higher than that in normal cells, providing an intelligent release mechanism for the cross-linked micelles with the disulfide bonds.17
As we know, vitamin analogue lipoic acid (LA) is an endogenous and hydrophobic substance, which has a disulfide-containing lipoic ring.18 Furthermore, LA molecules were successfully applied to the preparation of core crosslinked micelles for intelligent drug delivery system.19 The cross-linked mechanism is that the disulfide-containing lipoic ring of LA was subjected to ring-opening polymerization by thiol–disulfide exchange in the presence of a catalytic amount of dithiothreitol (DTT), resulting in the formation of stable linear polydisulfide crosslinks between different lipoyl units.20–22 Based on the hydrophobicity and bioactive efficacy of LA, LA-based polymeric amphiphiles were applied as delivery vehicles for hydrophobic anticancer agents.
An ideal nanocarrier should be non-toxic and biodegradable. Hydrophilic glycol chitosan (GC) is a derivative of chitosan with good biodegradability and low immunogenicity. Amphiphilic glycol chitosan derivatives, such as glycol chitosan bearing 5β-cholanic acid and cholesterol-modified glycol chitosan, have been reported as efficient carriers for gene and drug.23–27 Based on the above considerations, we attempted to use glycol chitosan as hydrophilic moieties to fabricate stimuli-responsive micelles.
In this study, the objective is to develop stimuli-responsive core-crosslinked nanocarriers. We synthesized glycol chitosan–lipoic acid (GC–LA) conjugate and prepared core-crosslinked GC–LA (GC–LA/cc) micelles by the catalysis reagent DTT. The anticancer drug doxorubicin (DOX), a typical cytotoxic anthracycline antibiotic, was selected as a model drug. DOX-loaded GC–LA/cc micelles were prepared and characterized. The construction of DOX-loaded GC–LA/cc micelle and its response to the endogenous high GSH in tumor cells were depicted in Scheme 1. We further investigated DOX release from drug-loaded micelles in vitro. The reduction sensitivity of DOX-loaded micelles was studied in vitro under different reduction conditions. In vitro anti-tumor cytotoxicity and intracellular uptake of DOX-loaded micelles were also evaluated in human lung adenocarcinoma A549 cells. Furthermore, the redox-responsive properties of DOX-GC–LA/cc micelles in tumor were carried out in tumor cells.
 |
| | Scheme 1 Illustration of the preparation of doxorubicin-loaded GC–LA (DOX-GC–LA) micelle and doxorubicin-loaded core-crosslinked GC–LA (DOX-GC–LA/cc) micelle, and schematic representation of the response to the endogenous high GSH in tumor cells of DOX-GC–LA/cc micelle. | |
2 Materials and methods
2.1 Materials
Glycol chitosan (Mw = 4.3 × 105, 75.2% of degree of deacetylation) and potassium poly(vinyl sulfate) were purchased from Wako Pure Chemical Industries, Ltd. (Osaka, Japan). Lipoic acid, 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC·HCl), dithiothreitol (DTT), glutathione (GSH), buthionine sulfoximine (BSO) and glutathione reduced ethyl ester (GSH-OET) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Pyrene was provided by Acros Organics (Beijing, China). Doxorubicin hydrochloride (DOX·HCl) was obtained from Beijing Huafeng United Technology Co, Ltd. (Beijing, China). Human lung adenocarcinoma (A549) cells were purchased from the Institute of Biochemistry and Cell Biology of Chinese Academy of Sciences (Shanghai, China). RPMI 1640 medium and trypsin–EDTA were obtained from Jinuo biotechnology company (Hangzhou, China). Fetal bovine serum (FBS) was from Sijiqing Biologic. Co. Ltd. (Zhejiang, China). 3-(4,5-Dimethyl-thiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) was purchased from Sigma-Aldrich (St. Louis, MO, USA). Hoechst 33342 was obtained from Beyotime Institute of Biotechnology (Haimen, China). All other chemicals were of analytical grade.
2.2 Synthesis of GC–LA conjugate
GC–LA conjugate was synthesized by covalent coupling of glycol chitosan and lipoic acid in the presence of EDC. Briefly, GC (720 mg, 1.67 μmol) was dissolved in 60 mL of deionized water, and ethanol (90 mL) was slowly added under magnetic stirring. Then, EDC·HCl (167.2 mg, 0.87 mmol) was added. After 1 h, 72 mg of lipoic acid (0.35 mmol) dissolved 46 mL ethanol were introduced into the above solution. This mixed solution was stirred to react for 24 h under nitrogen gas protection at 37 °C. Next, this solution was dialyzed (MWCO: 14 kDa) against deionized water for 24 h and filtered to remove the byproducts. The resultant solution was freeze-dried, and the product GC–LA was obtained.
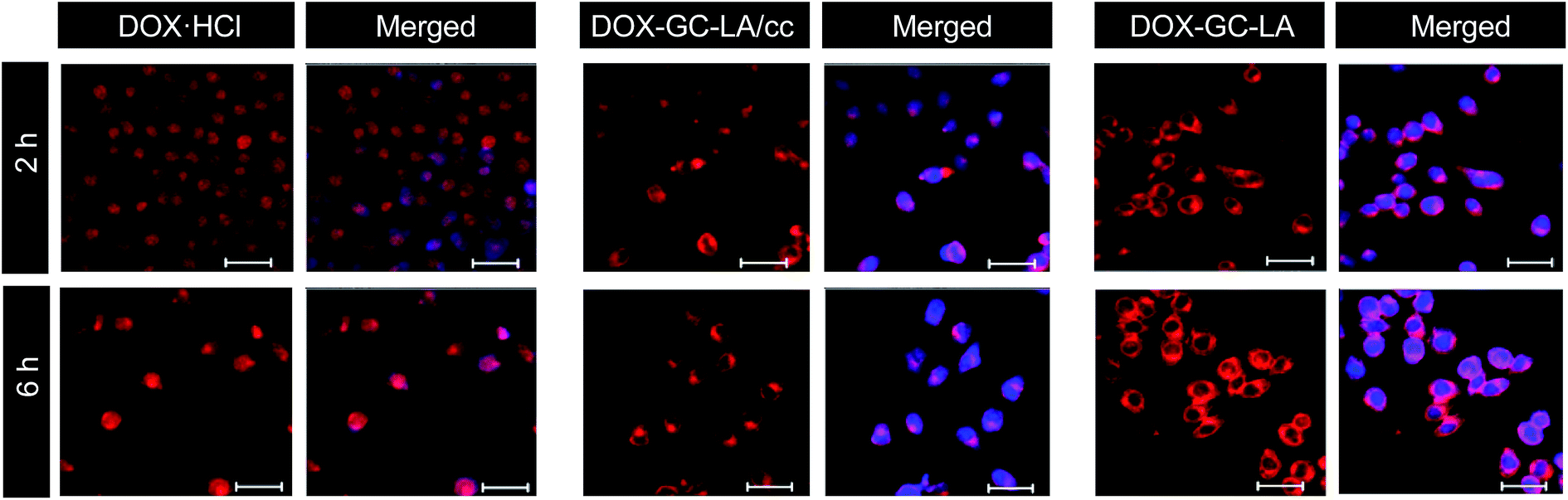
The chemical structure of GC–LA conjugate was confirmed by FTIR and 1H NMR. The IR spectrum of GC–LA was obtained as KBr pellets on FTIR spectrometer (Vertex 70, Bruker Corporation, Ettlingen, Germany). 1H NMR spectra of lipoic acid, GC and GC–LA were analyzed by using a NMR spectrometer (Avance DMX500, Bruker Corporation, Ettlingen, Germany). GC and GC–LA were dissolved in D2O, and lipoic acid was in CDCl3. The degree of substitution (DS) was expressed as the number of lipoic acid per 100 sugar residues of glycol chitosan, and determined by colloid titration assay as previously described.25,28 The method was based on the reaction between positively charged polyelectrolytes and negatively charged ones. Briefly, GC–LA (5 mg) or glycol chitosan (5 mg) were dissolved in aqueous 2% acetic acid solution (5 mL). 2.5 mM potassium poly(vinyl sulfate) solution was used as titrant. Toluidine blue (0.1%, w/v) was the indicator. The degree of deacetylation (DD) of glycol chitosan was firstly calculated using the eqn (1)–(4).28
| | |
X1 = 2.5 × 10−3 × 205 × f × ΔV1
| (1) |
| |
 | (3) |
where Δ
V1 is the net consumed volume (mL) of 2.5 mM PVSK solution, and
f is the concentration factor of PVSK solution.
X1 is the mass of glucosamine units contained in the sample of 5 mg glycol chitosan.
Y1 is the mass of acetyl glucosamine units contained in the sample of 5 mg glycol chitosan. DA is the degree of acetylation of glycol chitosan. As a result, DD and DA is 75.2% and 24.8%, respectively.
The number of LA groups per 100 sugar residues of glycol chitosan was calculated using the eqn (5)–(9).
| | |
X2 = 2.5 × 10−3 × 205 × f × ΔV2
| (5) |
| |
 | (6) |
| |
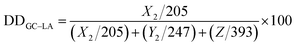 | (8) |
| | |
DS = 100 − DAGC–LA − DDGC–LA
| (9) |
where
X2 is the mass of
N-unsubstituted glucosamine units contained in the sample of 5 mg GC–LA.
Y2 is the mass of acetyl glucosamine contained in the sample of 5 mg GC–LA.
Z is the mass of
N-substituted glucosamine units contained in the sample of 5 mg GC–LA. Δ
V2 is the net consumed volume (mL) of 2.5 mM PVSK solution. DD
GC–LA is the number of glucosamine groups per 100 sugar residues of GC–LA. DA
GC–LA is the number of acetyl glucosamine groups per 100 sugar residues of GC–LA.
2.3 Preparation of blank core-crosslinked micelles
Blank core-crosslinked GC–LA micelles were prepared by two steps. First, blank GC–LA micelles were prepared by probe sonication in aqueous medium. GC–LA (200 mg) was dispersed in 100 mL water at room temperature for 12 h, followed by sonication using a probe type Sonifier (Scientz-IID, Ningbo Scientz Biotechnology Co. Ltd) at 200 W for 8 min in an ice bath. Second, GC–LA nanoparticle suspension was adjusted to pH 8.4 by borate buffer, and stirred to react with DTT for 24 h in nitrogen gas at 37 °C. The amount of DTT is 10 mol% relative to the content of lipoyl units in the micelles.19 Then, the mixed solution was dialyzed (MWCO: 14 kDa) against deionized water for 24 h to remove the impurities. The formation of crosslinked GC–LA micelles was evaluated by an UV-vis spectrophotometer (TU-1901, Beijing Purkinje General Instrument Co., Ltd, China). GC–LA or GC–LA/cc micelles were dissolved in deionized water at the concentration of 1 mg mL−1. Then, UV-vis spectrophotometer was used to scan the spectra of these solutions in the wavelength range of 200–600 nm.
2.4 Preparation of DOX-loaded cross-linked micelles
The non-crosslinked GC–LA micelles encapsulating DOX were prepared by the o/w method. GC–LA micelles (2 mg mL−1) were prepared as described above. DOX·HCl was separately dissolved in chloroform with 3 equivalent molar ratio of triethylamine, and stirred overnight under the dark condition. Then, different amount of DOX (10, 15 or 20 mg) in organic phase solution was added dropwise to 50 mL aqueous phase under vigorously stirring to form the o/w emulsion. Chloroform was evaporated by using rotary evaporator under reduced pressure. The solution was placed into the dialysis bag (MWCO: 14 kDa) for dialysis against deionized water for 24 h to remove free DOX. And almost no DOX can be detected in the dialyzed water by fluorescence spectroscopy. Afterwards, the DOX-loaded micelles (DOX-GC–LA) was prepared.
Next, DOX-GC–LA micelles were adjusted to pH 8.4 by borate buffer and stirred to react with DTT for 24 h as stated above. The mixed solution was dialyzed against deionized water for 24 h. Finally, DOX-GC–LA and DOX-GC–LA/cc micelles were lyophilized.
2.5 Characterization of blank and drug-loaded micelles
Fluorescence spectroscopy was adopted to analyze the critical micelle concentration (CMC) of GC–LA. Briefly, the GC–LA solution was diluted to various concentrations by deionized water. 1 mL of pyrene in acetone was separately added to 10 mL flasks, and the solvent was evaporated at 40 °C. Next, 10 mL of various concentrations of sample solution were added to each flask, and heated at 50 °C for 12 h to equilibrate the pyrene and the micelles. The solution remained undisturbed to cool overnight at room temperature. The final concentration of pyrene was 6.0 × 10−7 M. Steady-state fluorescent spectra were measured by fluorescence spectrophotometer (Perkin-Elmer LS55, Perkin-Elmer Ltd., Llantrisant, UK). The slit width is 10 nm. The emission and excitation wavelength was set at 390 and 339 nm, respectively.
The sizes of blank and drug-loaded micelles were detected by dynamic light scattering (DLS) using a Zetasizer (90Plus, Brookhaven Instruments Corp., New York, NY, USA). The morphology was observed by transmission electron microscopy (TEM, JEM-1230, Jeol, Tokyo, Japan). The sample was placed onto a 300-mesh copper grid coated with carbon and the extra solution was blotted with filter paper. Further the sample was stained with 2.0% (w/v) phosphotungstic acid and followed by air-drying. Observation was done at 80 kV.
The drug loading content (LC) and encapsulation efficiency (EE) of DOX-loaded micelles were determined by extracting DOX from the micelles with dimethyl sulphoxide (DMSO) and using UV method at 480 nm. The LC and EE were calculated by the following equations:
| |
 | (10) |
| |
 | (11) |
2.6 Reduction-triggered destabilization of DOX-loaded crosslinked micelles
The aim of this part was to study the reduction sensitivity of DOX-GC–LA/cc micelles. The size changes of DOX-GC–LA/cc micelles in response to different reduction environments were determined by DLS. Briefly, DOX-GC–LA/cc lyophilates were dissolved in phosphate buffered saline (PBS, pH 7.4) and followed by probe sonication. Then, known amounts of GSH were added to the above DOX-GC–LA/cc micelles (2 mg mL−1). The final GSH concentrations were 0, 10 μM, 3.5 mM, or 20 mM. These solutions were placed at 37 °C and 100 rpm in the air-bath shaker. The sizes of these micelles were detected at predetermined time intervals. All samples were in triplicates.
2.7 Reduction-triggered release of DOX-GC–LA/cc micelles
The release of DOX from DOX-GC–LA and DOX-GC–LA/cc micelles was studied in vitro by a dialysis method in phosphate buffered saline (PBS, pH 7.4) or PBS (pH 7.4) containing 20 mM GSH. Briefly, 1 mL DOX-loaded micelles was placed in a dialysis bag (MWCO: 14 kDa) and dialyzed against 20 mL of release medium. The condition was set at 37 °C and maintained at 160 rpm in an air-bath shaker. The entire media outside the dialysis bag were collected and replaced with 20 mL of fresh media at appropriate periods. The amount of DOX in release medium was determined by fluorescence spectrophotometer (Perkin-Elmer LS55, Perkin-Elmer Ltd., Llantrisant, UK), which applied an excitation wavelength of 470 nm and an emission wavelength of 585 nm. The release experiments were performed in triplicate.
2.8 Cell culture
Human lung adenocarcinoma (A549) cells were routinely cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin–streptomycin solution. The cells were maintained at 37 °C in a humidified atmosphere containing 5% CO2.
2.9 Intracellular uptake study
Confocal Laser Scanning Microscopy (CLSM) was used to study the cellular uptake of DOX-loaded micelles. Specifically, A549 cells were cultured at a density of 2.0 × 105 cells per well in 6-well plates (Costar, Corning, NY, USA) for 24 h. After removed the cultured media, DOX·HCl, DOX-GC–LA or DOX-GC–LA/cc micelles (equivalent DOX concentration: 10 μg mL−1) were added. After 2 or 6 h incubation, the cells were washed two times with PBS (pH 7.4) and fixed in 4% paraformaldehyde solution for 30 min. Further, the cells were treated with Hoechst 33342 (5 μg mL−1) for nuclei staining. After 15 min, A549 cells were washed with PBS. The cells were examined by a Zeiss LSM-510 confocal microscope (Carl Zeiss LSM-510, Germany) under the identical settings.
The GSH concentration in tumor cells could be regulated by pretreatment with GSH-OET and BSO.15 The effect of cellular uptake from DOX-GC–LA/cc micelles was studied in different reduction environment. A549 cells were seeded as described above for 24 h, and then incubated with 20 mM GSH-OET for 4 h or 1.0 mM BSO for 12 h. Then, the DOX-GC–LA/cc micelles including 10 μg mL−1 DOX were added. Untreated cells were used as a control. After 6 h incubation, a series of treatment were made as mentioned above. A549 cells were observed by using the Zeiss LSM-510 confocal microscope.
2.10 In vitro cytotoxicity
The cytotoxicity of blank and drug-loaded micelles was evaluated by using MTT assay. A549 cells were seeded at a density of 1.0 × 104 cells per well in 96-well plates (Costar, Corning, NY, USA) and allowed to grow overnight. After removed the cultured media, a series of concentrations of blank and DOX-encapsulated micelles were added and incubated for 48 h. Then 30 μL MTT solution (5 mg mL−1) was added and treated for 4 h. Afterward, the solution was aspirated, and the formazan crystals in live cells were solubilized with 200 μL of DMSO for 15 min. The absorbance was measured at 570 nm using a microplate reader (Thermo Scientific Multiskan MK3, Hudson, USA).
In order to validate that the cytotoxicity was related to the intracellular GSH-responsive ability of DOX-GC–LA/cc micelles. GSH-OET and BSO were used as external GSH enhancer and reducer, respectively. Typically, A549 cells were seeded and allowed to be adhere as described above. The cells were incubated with 20 mM GSH-OET for 4 h or 1.0 mM BSO for 12 h. Then the cells were washed twice with PBS to remove GSH-OET or BSO in the medium and further added with DOX-GC–LA/cc micelles. The final concentration of DOX is 5 or 10 μg mL−1. After 24 or 48 h, the cells were done as previously stated. The absorbency values were measured by microplate reader (Thermo Scientific Multiskan MK3, USA).
2.11 Statistics analysis
Data are expressed as mean ± standard deviation (SD). One-way analysis of variance (ANOVA) was used for data analysis. P < 0.05 was considered significant. Extreme significance was set at P < 0.01.
3 Results and discussion
3.1 Synthesis and characterization of GC–LA conjugate
The synthesis of GC–LA was illustrated in Fig. 1. The coupling reagent EDC is called “zero length” cross-linker because the amide linkages are developed without leaving a spacer molecule. GC–LA was synthesized by the formation of amide bonds between carboxyl groups of lipoic acid and amino groups of glycol chitosan. The FTIR spectra of GC and GC–LA were shown in Fig. S1.† As described above, the synthetic reaction of lipoic acid conjugated glycol chitosan was actually a process that the content of amide bond increased, while the level of primary amino group decreased. The characteristic peak of amide linkage in glycol chitosan was observed at 1647 cm−1, whereas this peak of GC–LA was appeared at 1665 cm−1. The phenomenon is similar to the report described by Zhang et al.29 The conjugation of LA with glycol chitosan was further confirmed by 1H NMR spectra (Fig. 2). Compared with glycol chitosan, new proton peaks in the GC–LA copolymer were observed at 1.2–1.6, 2.0–2.4 and 3.2 ppm.21 The peaks were attributed to LA groups. This result indicated that GC–LA conjugate was successfully synthesized. According to the colloidal titration method, the degree of substitution was 8.3 LA groups per 100 sugar units of glycol chitosan.
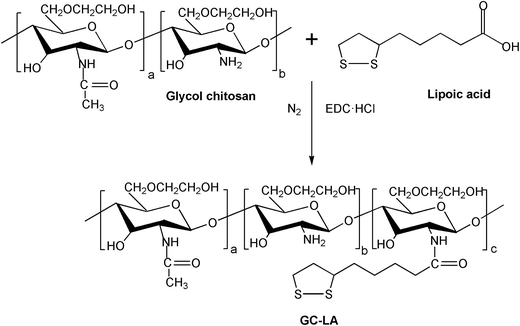 |
| | Fig. 1 Synthetic scheme of glycol chitosan–lipoic acid conjugate (GC–LA). | |
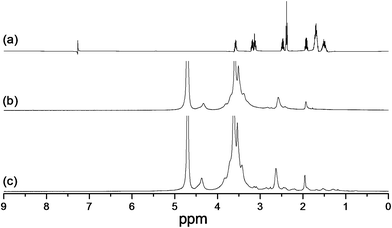 |
| | Fig. 2 1H nuclear magnetic resonance spectra of (a) lipoic acid, (b) glycol chitosan and (c) GC–LA. | |
3.2 Preparation and characterization of blank micelles
The CMC of GC–LA conjugate was determined by fluorescence spectra using pyrene as a hydrophobic fluorescence probe.30 The CMC is assumed to occur where there is a sharp increase in the ratio of the fluorescence intensities (I338/I333). Fig. 3 showed the intensity ratio of I338/I333 vs. log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) C of GC–LA conjugate for the pyrene excitation spectra, resulting in confirming the CMC obtained from the intersection of two straight lines. The CMC of GC–LA was 0.081 mg mL−1. The result indicated that the GC–LA copolymer can be easy to form micelles even under the diluted solution.
C of GC–LA conjugate for the pyrene excitation spectra, resulting in confirming the CMC obtained from the intersection of two straight lines. The CMC of GC–LA was 0.081 mg mL−1. The result indicated that the GC–LA copolymer can be easy to form micelles even under the diluted solution.
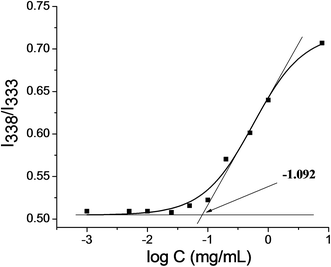 |
| | Fig. 3 Plot of the intensity ratio I338/I333 from pyrene excitation spectra of GC–LA as a function of logC. | |
Lipoic acid-based copolymers can form core-crosslinked micelles in aqueous media under the catalysis of DTT. As shown in Fig. S2,† the UV-vis spectra of blank GC–LA micelles had the absorbance at 330 nm, whereas this absorbance was disappeared in the core-crossed GC–LA micelles. This result was due to the fracture of the disulfide bond within the lipoyl ring and the formation of linear polydisulfide bonds between adjacent LA units.20 Therefore, DTT had successfully promoted the core-crosslinking of GC–LA nanoparticles.
In addition, the mean size of blank GC–LA micelles determined by DLS was 287 nm, while that of crosslinked counterpart was 281 nm. It was found that these micelles had nearly identical sizes, indicating that the cross-linkages among the LA moieties occur within the micelles core. And the crosslinking has little influence on the hydrodynamic diameter of the micelles.31
3.3 Preparation and characterization of DOX-loaded micelles
DOX-loading of non-crosslinked GC–LA micelles was adopted by an oil-in-water emulsion method. Subsequently, these DOX-loaded micelles were readily cross-linked with a catalytic amount of DTT. The physicochemical properties of drug-loaded micelles are summarized in Table 1. As the feed ratios of DOX to carrier (1/10, 1.5/10, 2/10, w/w) increased, drug loading content and mean size of DOX-loaded micelles increased. The mean diameters of DOX-GC–LA were in the range of 312–413 nm, and those of the crosslinked counterparts (DOX-GC–LA/cc) increased from 305 nm to 408 nm. These results demonstrated that the particle size of DOX-loaded micelles was larger than that of their blank polymeric micelles. It was possibly due to the fact that DOX molecules were loaded into the micelles and the inner space of these micelles increased. Moreover, it was found that the mean sizes of DOX-GC–LA micelles were almost identical with the crosslinked counterparts encapsulating DOX. It implied that crosslinking behaviors had little effect on the sizes of DOX-loaded micelles.
Table 1 Physicochemical characteristic of DOX-loaded micelles
| Sample |
Drug/carriera |
LCb (%) |
EEc (%) |
Sized (nm) |
PIe |
| The ratio of DOX to carrier, based on feed amount (mg mg−1). Loading content. Encapsulation efficiency. Measured by dynamic light scattering. Polydispersity index. |
| DOX-GC–LA-1 |
1/10 |
6.94 |
74.60 |
312 ± 28.4 |
0.241 ± 0.014 |
| DOX-GC–LA-1/cc |
1/10 |
6.03 |
64.17 |
305 ± 13.1 |
0.163 ± 0.027 |
| DOX-GC–LA |
1.5/10 |
10.50 |
78.19 |
349 ± 20.9 |
0.219 ± 0.025 |
| DOX-GC–LA/cc |
1.5/10 |
9.65 |
71.19 |
335 ± 30.7 |
0.167 ± 0.034 |
| DOX-GC–LA-2 |
2/10 |
11.82 |
67.04 |
413 ± 20.3 |
0.185 ± 0.011 |
| DOX-GC–LA-2/cc |
2/10 |
10.74 |
60.16 |
408 ± 15.5 |
0.258 ± 0.028 |
As the ratio of drug to carrier was 1.5/10, the entrapment efficiency of DOX-GC–LA micelles was 78.19%, which is higher than that of other DOX-loaded micelles. Considering the particle size, the drug loading property and the entrapment efficiency, the optimal ratio of drug to carrier (1.5/10) was chosen to prepare DOX-loaded micelles. The DOX-GC–LA (DOX loading: 10.50%) and DOX-GC–LA/cc (DOX loading: 9.65%) micelles named was selectively used for the following studies.
The particle morphology of DOX-GC–LA/cc micelles was observed by a transmission electron microscopy (TEM). The TEM image depicted that the micelles were almost spherical in shape (Fig. 4). It was obviously found that the sizes of these micelles were smaller than that determined by DLS. This result is possibly due to the sample preparation process that TEM analysis was in the dried state, while DLS sample is in the hydrated state.32
 |
| | Fig. 4 Transmission electron microscopy image of DOX-GC–LA/cc micelles. | |
3.4 In vitro reduction-sensitivity of DOX-loaded micelles
To study the reduction sensitivity of DOX-GC–LA/cc micelles in vitro, DLS was employed to detect the variety of the micelle's size at predetermined time points. The different GSH content in PBS solution was used to simulate the conditions of the blood plasma, normal cells and tumor cells. As presented in Fig. 5, the particle size of DOX-GC–LA/cc remained almost unchanged in PBS with 0 or 10 μM of GSH during 48 h incubation. After treatment with 3.5 mM GSH-containing PBS (pH 7.4), the diameter of these micelles turned larger from 329 nm to 751 nm. When the crosslinked micelles were incubated with 20 mM GSH PBS (pH 7.4), the diameter rapidly increased from 329 nm to 1334 nm. The size distributions of these particles determined by DLS were provided in Fig. S3.† These results revealed that the polydisulfide crosslinks within the GC–LA/cc could be rapidly reduced and sabotaged by the cancer intracellular level of GSH, leading to the dissociation of the micelles and the disclosure of drugs.
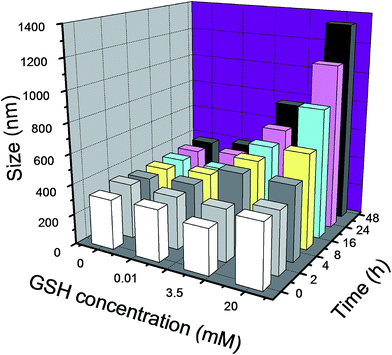 |
| | Fig. 5 Size changes of DOX-GC–LA/cc micelles in PBS (pH 7.4) containing 0, 10 μM, 3.5 mM or 20 mM GSH at 37 °C for 48 h. Data are presented as the average (n = 3). | |
In order to appraise the potential of these nanoparticles as drug carriers, the release behavior of DOX from drug-loaded micelles was studied in PBS at pH 7.4, simulating the in vivo biological environment. As shown in Fig. 6, DOX release from DOX-GC–LA/cc was slow at the early time points. There was nearly 12.3% of drug release from crosslinked micelles over 12 h, while DOX-GC–LA micelles released 35.3% of DOX over the same period. What's more, the cumulative release of DOX from the crosslinked micelles was only about 25.3% at 96 h, which was markedly lower (P < 0.01) than that released from the non-crosslinked micelles (87.6%). Although the crosslinking core structure has no enough capacity to completely block drug release from the micelles in PBS, the polydisulfide crosslinks can strengthen the micellar structure and allow the crosslinked micelles to better reserve hydrophobic drug molecules within the micellar core. In addition, we also performed the release study of DOX-GC–LA/cc micelles in PBS containing 20 mM GSH corresponding to the cancer intracellular GSH level. A surprising finding was that about 45.5% of DOX was released from the crosslinked micelles at 12 h, and full release of DOX emerged at 96 h, which was even faster than that of DOX-GC–LA micelles in the absence of GSH. We inferred that polydisulfide linkage in the crosslinking core structure can rapidly disconnect in the reduction condition within the cancer cells. As a consequence, the generated free thiols could greatly decrease the hydrophobic interactions inside the micellar core, leading to the accelerated drug release. Additionally, the cumulative release of DOX from DOX-GC–LA micelles was 90.1% for 96 h in the presence of 20 mM GSH, which is slightly higher (P > 0.05) than that of DOX-GC–LA in the absence of 20 mM GSH. We inferred that reduced GSH had not significant influence on the drug release of DOX-GC–LA micelles. These results suggested that the core-crosslinked micelles would provide with decreasing premature drug release in blood and increasing selectively drug release in tumor.
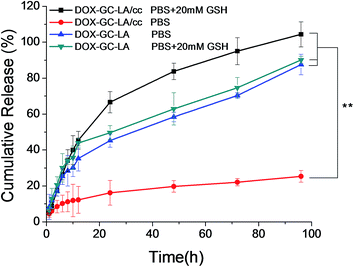 |
| | Fig. 6 Release profiles of DOX from DOX-loaded micelles in PBS (pH 7.4) or PBS (pH 7.4) including 20 mM GSH at 37 °C for 96 h. All results show representative data obtained from three independent experiments. **P < 0.01. | |
3.5 Intracellular uptake study
The intracellular uptake of DOX·HCl or DOX-loaded micelles was investigated by CLSM in A549 cells. As shown in Fig. 7, the red fluorescence was emitted from DOX formulations and the blue fluorescence stained in nuclei was from Hoechst 33342. After incubation for 2 and 6 h, the red fluorescence from DOX·HCl was seen mostly in the nuclei, while DOX fluorescence from DOX-loaded micelles was also distributed mainly in cytoplasm. The different result was interpreted that the free drug (DOX·HCl) could rapidly enter the cancer cells by passive diffusion, while DOX-loaded micelles were transported into the cells by energy-dependent endocytosis manner, leading to a relatively slow rate into the tumor cells.33 Additionally, DOX-loaded micelles were not easy to directly transport into the nuclei. This is because of the large particle size and high molecular weight of DOX-loaded micelles.34 We also found that the red fluorescence emitted from DOX-GC–LA micelles was stronger than that of DOX-GC–LA/cc micelles after incubation for 2 h, and maintained the superiority with further incubation for 6 h. We inferred that crosslinked disulfide linkage hampered the DOX release from DOX-GC–LA/cc micelles, and the breakage of core crosslinked structure was a time-consuming process.
 |
| | Fig. 7 CLSM images of A549 cells after incubation with DOX·HCl, DOX-GC–LA/cc micelles and DOX-GC–LA micelles for 2 and 6 h. Hoechst 33342 staining the nucleus showed blue fluorescence. The scale bars correspond to 50 μm in all the images. | |
We are also interested in the redox-responsive properties of DOX-GC–LA/cc micelles in living cells. It is well known that GSH-OET is an intracellular GSH enhancer, and BSO is an inhibitor for the intracellular synthesis of GSH.35 A549 cells were treated with GSH-OET or BSO, and then incubated with the DOX-loaded crosslinked micelles. Meanwhile, the non-pretreated cells were used as control. CLSM images show that the intracellular DOX fluorescence in the GSH-OET pretreated cells is higher than the control cells (Fig. 8). Moreover, the fluorescence intensity in A549 cells was markedly decreased after the BSO pretreatment. These results indicated that pretreated with inhibitor and accelerator of GSH had an impact on the intracellular GSH levels and the DOX release rates of drug-loaded crosslinked micelles.
 |
| | Fig. 8 The CLSM images of BSO pretreated, GSH-OET pretreated and untreated A549 cells after the cells were incubated with DOX-GC–LA/cc micelles for 6 h. Hoechst 33342 staining the nucleus showed blue fluorescence. The scale bar is 50 μm. | |
3.6 In vitro cytotoxicity
In vitro cytotoxic activity of blank and DOX-loaded micelles against A549 cells was investigated. As shown in Fig. 9a, blank GC–LA and GC–LA/cc micelles were almost non-toxic (cell viabilities >90%) up to a tested concentration of 1 mg mL−1. These results indicated that these copolymers had the characteristics of safety and biocompatibility. Subsequently, the cytotoxicity of DOX·HCl, DOX-GC–LA and DOX-GC–LA/cc was examined in A549 cells at DOX concentration ranging from 0.01 to 10 μg mL−1 for 48 h (Fig. 9b). The cytotoxicity studies demonstrated that both DOX-loaded micellar formulations revealed lower cytotoxicity than DOX·HCl. Similar results were reported by Zhong group.19,22 The IC50 values of DOX·HCl, DOX-GC–LA and DOX-GC–LA/cc micelles were 0.19, 0.52, and 0.63 μg mL−1, respectively. This result was in line with the CLSM observation. DOX-loaded micelles accumulated in the nucleus were less than DOX·HCl. This is due to the fact that DOX·HCl can be quickly transported into cells by passive diffusion, while DOX-loaded micelles were internalized by endocytosis. Moreover, DOX released from the crosslinked micelles consumed more time than that from non-crosslinked micelle. Thus, DOX-GC–LA/cc micelles exhibited lower cytotoxicity than DOX-GC–LA micelles. Moreover, it was known that drug-loaded micelles can efficiently deliver the drug to tumor cells and improve the anti-tumor activity by EPR effect. We deduced that the DOX-GC–LA/cc micelles could effectively prevent premature drug release in blood and increase DOX concentration in the tumor sites.
 |
| | Fig. 9 The in vitro cytotoxicity of (a) blank GC–LA and GC–LA/cc micelles and (b) DOX·HCl, DOX-GC–LA and DOX-GC–LA/cc micelles against A549 cells after 48 h incubation. | |
In order to further validate the reduction-triggered intracellular DOX release, the cell viability of DOX-GC–LA/cc micelles was investigated in GSH-OET pretreated, BSO pretreated and untreated A549 cells. As shown in Fig. 10, DOX-GC–LA/cc micelles in BSO pretreated cells exhibited significantly decreased cell inhibition than the micelles in untreated A549 cells at 24 h (DOX concentration is 5 and 10 μg mL−1). Meanwhile, these micelles showed improved inhibition after the cells were pretreated with GSH-OET. The difference of cell viability was attributed to the enhancement of intracellular GSH levels by GSH-OET and the decease of GSH concentrations with BSO. Thus it leaded to regulate the drug release and DOX anticancer activity. This result agreed with CLSM observation. Furthermore, the phenomenon can be observed as DOX-GC–LA/cc micelles were incubated with the cells for 48 h as the DOX concentration is 5 μg mL−1. However, the cell viability of DOX-GC–LA/cc micelles exhibited almost equal in the GSH-OET pretreated, BSO pretreated and untreated A549 cells after 48 h incubation (DOX concentration: 10 μg mL−1). This is possibly due to the fact that high DOX concentration and prolonged treatment time could produce high cell mortality. Then the effects of GSH-OET or BSO could not be observed in this experiment. The above results indicated that the crosslinked biodegradable micelles had the potential as intelligent drug delivery vehicles.
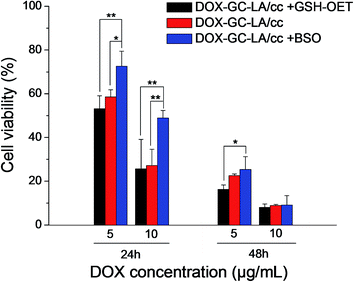 |
| | Fig. 10 Cell viability ratio changes of the GSH-OET pretreated, BSO pretreated and untreated A549 cells after the cells were incubated with DOX-GC–LA/cc micelles for 24 h or 48 h. *P < 0.05, **P < 0.01. | |
4 Conclusion
In this work, GC–LA copolymer was successfully synthesized and characterized. It can self-assemble into nanoscaled micelles in aqueous medium and be further crosslinked by a catalytic amount of DTT. These nanoparticles show relatively high DOX-loading capacity and low cytotoxicity themselves. In addition, the core crosslinked GC–LA micelles are able to retain excellent reduction sensitivity in the cancer intracellular environment, which probably enhance the delivery of DOX to tumor tissues. Taken together, these reduction-responsive core-crosslinked micelles could be used as drug carriers for cancer therapy.
Acknowledgements
We appreciate the financial support from National Natural Science Foundation of China (No. 81360484), Natural Science Foundation of Jiangxi Province (No. 20151BAB205081), Scientific Research Fund of Jiangxi Health Department (No. 20157105).
References
- Y. Miura, T. Takenaka, K. Toh, S. Wu, H. Nishihara, M. R. Kano, Y. Ino, T. Nomoto, Y. Matsumoto, H. Koyama, H. Cabral, N. Nishiyama and K. Kataoka, ACS Nano, 2013, 7, 8583–8592 CrossRef CAS PubMed.
- K. Byungkuk, L. Eunsun, K. Yerang, P. Sanga, G. Khang and L. Dongwon, Adv. Funct. Mater., 2013, 23, 5091–5097 CrossRef.
- J. Yu, X. Xie, X. Xu, L. Zhang, X. Zhou, H. Yu, P. Wu, T. Wang, X. Che and Z. Hu, J. Mater. Chem. B, 2014, 2, 2114–2126 RSC.
- H. Yu, Z. Cui, P. Yu, C. Guo, B. Feng, T. Jiang, S. Wang, Q. Yin, D. Zhong, X. Yang, Z. Zhang and Y. Li, Adv. Funct. Mater., 2015, 25, 2489–2500 CrossRef CAS.
- S. J. Yu, C. L. He, Q. Lv, H. Sun and X. S. Chen, RSC Adv., 2014, 4, 63070–63078 RSC.
- E. S. Lee, J. H. Kim, T. Sim, Y. S. Youn, B.-J. Lee, Y. T. Oh and K. T. Oh, J. Mater. Chem. B, 2014, 2, 1152–1159 RSC.
- J. Yu, Y. Zhou, W. Chen, J. Ren, L. Zhang, L. Lu, G. Luo and H. Huang, Materials, 2015, 8, 6685–6696 CrossRef.
- L. Y. Qiu and M. Q. Yan, Acta Biomater., 2009, 5, 2132–2141 CrossRef CAS PubMed.
- A. B. E. Attia, P. Oh, C. Yang, J. P. K. Tan, N. Rao, J. L. Hedrick, Y. Y. Yang and R. Ge, Small, 2014, 10, 4281–4286 CAS.
- Q. Hu, C. J. Rijcken, R. Bansal, W. E. Hennink, G. Storm and J. Prakash, Biomaterials, 2015, 53, 370–378 CrossRef CAS PubMed.
- H. S. Han, K. Y. Choi, H. Ko, J. Jeon, G. Saravanakumar, Y. D. Suh, D. S. Lee and J. H. Park, J. Controlled Release, 2015, 200, 158–166 CrossRef CAS PubMed.
- R. R. Wakaskar, S. P. R. Bathena, S. B. Tallapaka, V. V. Ambardekar, N. Gautam, R. Thakare, S. M. Simet, S. M. Curran, R. K. Singh, Y. Dong and J. A. Vetro, Pharm. Res., 2015, 32, 1028–1044 CrossRef CAS PubMed.
- H. S. Han, T. Thambi, K. Y. Choi, S. Son, H. Ko, M. C. Lee, D.-G. Jo, Y. S. Chae, Y. M. Kang, J. Y. Lee and J. H. Park, Biomacromolecules, 2015, 16, 447–456 CrossRef CAS PubMed.
- L. L. Gao, Q. J. Luo, Y. Wang, H. Du, X. D. Li, Z. Q. Shen and W. P. Zhu, RSC Adv., 2014, 4, 4177–4180 RSC.
- Y. W. Hu, Y. Z. Du, N. Liu, X. Liu, T. T. Meng, B. L. Cheng, J. B. He, J. You, H. Yuan and F. Q. Hu, J. Control Release, 2015, 206, 91–100 CrossRef CAS PubMed.
- G. Saito, J. A. Swanson and K. D. Lee, Adv. Drug Delivery Rev., 2003, 55, 199–215 CrossRef CAS PubMed.
- S. Joshi-Barr, C. D. G. Lux, E. Mahmoud and A. Almutairi, Antioxid. Redox Signaling, 2014, 21, 730–754 CrossRef CAS PubMed.
- Y.-W. Won, S.-M. Yoon, K. S. Lim and Y.-H. Kim, Adv. Funct. Mater., 2012, 22, 1199–1208 CrossRef CAS.
- Y.-L. Li, L. Zhu, Z. Liu, R. Cheng, F. Meng, J.-H. Cui, S.-J. Ji and Z. Zhong, Angew. Chem., Int. Ed., 2009, 48, 9914–9918 CrossRef CAS PubMed.
- W. Fan, Y. Wang, X. Dai, L. Shi, D. McKinley and C. Tan, Pharm. Res., 2015, 32, 1325–1340 CrossRef CAS PubMed.
- Y. Xu, F. Meng, R. Cheng and Z. Zhong, Macromol. Biosci., 2009, 9, 1254–1261 CrossRef CAS PubMed.
- R. Wei, L. Cheng, M. Zheng, R. Cheng, F. Meng, C. Deng and Z. Zhong, Biomacromolecules, 2012, 13, 2429–2438 CrossRef CAS PubMed.
- J. Yu, Y. Liu, L. Zhang, J. Zhao, J. Ren, L. Zhang and Y. Jin, J. Biomater. Sci., Polym. Ed., 2015, 26, 1475–1489 CrossRef CAS PubMed.
- J. M. Yu, Y. J. Li, L. Y. Qiu and Y. Jin, Eur. Polym. J., 2008, 44, 555–565 CrossRef CAS.
- S. Kwon, J. H. Park, H. Chung, I. C. Kwon, S. Y. Jeong and I. S. Kim, Langmuir, 2003, 19, 10188–10193 CrossRef CAS.
- H. Y. Hwang, I. S. Kim, I. C. Kwon and Y. H. Kim, J. Controlled Release, 2008, 128, 23–31 CrossRef CAS PubMed.
- N. Duhem, J. Rolland, R. Riva, P. Guillet, J.-M. Schumers, C. Jerome, J.-F. Gohy and V. Preat, Int. J. Pharm., 2012, 423, 452–460 CrossRef CAS PubMed.
- J. Yu, X. Xie, J. Wu, Y. Liu, P. Liu, X. Xu, H. Yu, L. Lu and X. Che, J. Biomater. Sci., Polym. Ed., 2013, 24, 606–620 CrossRef CAS PubMed.
- L. Zhang, J. Yao, J. Zhou, T. Wang and Q. Zhang, Int. J. Pharm., 2013, 441, 654–664 CrossRef CAS PubMed.
- J. Zhang and P. X. Ma, Angew. Chem., Int. Ed., 2009, 48, 964–968 CrossRef CAS PubMed.
- W. Chen, F. Meng, R. Cheng, C. Deng, J. Feijen and Z. Zhong, J. Controlled Release, 2015, 210, 125–133 CrossRef CAS PubMed.
- B. Khorsand, G. Lapointe, C. Brett and J. K. Oh, Biomacromolecules, 2013, 14, 2103–2111 CrossRef CAS PubMed.
- M. Li, Z. Tang, S. Lv, W. Song, H. Hong, X. Jing, Y. Zhang and X. Chen, Biomaterials, 2014, 35, 3851–3864 CrossRef CAS PubMed.
- J. Yu, X. Xie, M. Zheng, L. Yu, L. Zhang, J. Zhao, D. Jiang and X. Che, Int. J. Nanomed., 2012, 7, 5079–5090 CrossRef CAS PubMed.
- J. Liu, W. Huang, Y. Pang, P. Huang, X. Zhu, Y. Zhou and D. Yan, Angew. Chem., Int. Ed., 2011, 50, 9162–9166 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra05501j |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.