
Photoluminescence properties of transition metal-doped Zn–In–S/ZnS core/shell quantum dots in solid films
Jiaming Liab,
Yang Liub,
Jie Huab,
Lianhua Tian*a and
Jialong Zhao*b
aDepartment of Physics, Yanbian University, Yanji 133002, China. E-mail: lhtian@ybu.edu.cn
bKey Laboratory of Functional Materials Physics and Chemistry of the Ministry of Education, Jilin Normal University, Siping 136000, China. E-mail: zhaojl@ciomp.ac.cn
First published on 3rd May 2016
Abstract
The photoluminescence (PL) properties of transition metal ion (Mn2+ or Cu+) doped Zn–In–S/ZnS core/shell quantum dots (QDs) in solution and solid films were investigated by using steady-state and time-resolved PL spectra. The PL peaks and lifetimes of Mn or Cu doped Zn–In–S/ZnS QD films exhibited almost no variation in contrast to those of the corresponding QD/PMMA blend films, indicating that no energy transfer process occurred in the pure doped QD films, while an obvious energy transfer was found in CdSe/ZnS QD films. Compared with the QDs in solution, the PL lifetimes of pure Mn or Cu doped Zn–In–S/ZnS QD films dropped only 16.2–37.5% which was attributed to the formation of surface traps/defects due to the loss of ligands on the surface of QDs while that of CdSe/ZnS QD films was shortened to the initial value of 50% which mainly came from energy transfer between QDs. Furthermore, the change in PL lifetimes found in Mn or Cu doped QD films could be reduced effectively by increasing the shell thickness of QDs. The experimental results are beneficial to the design of Mn or Cu doped QD-based photoelectronic devices.
1. Introduction
In the past three decades, colloidal quantum dots (QDs) with high photoluminescence (PL) efficiency have been extensively investigated in many fields, such as biological imaging, solid-state lighting, and photovoltaic devices.1–6 Despite II–VI QDs possessing promising optoelectronic properties, the heavy metals such as Cd or Pb have potential adverse effects on the environment, which limits their validation in large-scale applications. Thus, more and more researchers are putting great effort into the development of cadmium-free transition metal ions Mn2+ or Cu+ doped ternary/quaternary alloyed QDs as an alternative to II–VI QDs, such as Cu:Zn–In–S,7,8 Mn:Cu–In–S,9,10 and Mn:Cu–Zn–In–S,11 Mn:Ag–Zn–In–S.12 These doped QDs exhibit high PL quantum yields (QYs), good thermal and chemical stability, larger Stokes shift, and longer PL lifetimes. Moreover, the absorption and emission properties for the doped QDs can be finely and widely engineered through controlling the host composition. These properties make them ideal for application in biotechnology and photoelectronic devices.12–16QDs-based light-emitting diodes (LEDs) have been widely studied as promising light sources because of their low scattering, high luminous efficiency and color rendering index (CRI) compared with conventional LEDs based on phosphors Y3A15O12:Ce3+.14,17,18 However, it is noteworthy that the QDs are needed to deposit onto substrates to form solid films for the fabrication of optoelectronic devices. Although some researchers have reported on the recombination mechanism in Mn or Cu doped QDs, the PL properties of the closely packed QDs are different from those of individual QDs due to different chemical environments, such as in solutions or solid films. Therefore, understanding the optical properties of doped QDs in solid films and properly controlling them are very important to design QDs-based optoelectronic devices. In this paper, we reported a systematic study of the steady-state and time-resolved PL behaviors of Mn or Cu doped Zn–In–S/ZnS core/shell QDs with different host composition in solution and solid films prepared by spin-coating or dropping method. The comparison of the optical properties between Mn (or Cu) doped Zn–In–S/ZnS QDs and CdSe/ZnS QDs is carried out to discuss the change in their PL emission peaks and lifetimes.
2. Experimental
2.1 Preparation of Cu (or Mn):ZnInS/ZnS and CdSe/ZnS core/shell QDs and QD films
In this experiment, the Cu:ZnInS/ZnS QDs were synthesized via a noninjection approach as described in ref. 7. The Cu:ZnInS cores were formed firstly and then followed by the growth of ZnS shell by introducing Zn precursors solution. The yellow (λ = 580 nm, PL QY = 47%) and red (λ = 620 nm, PL QY = 40%) Cu:ZnInS/ZnS core/shell QDs prepared with Zn/In precursor ratio of 1/2 and 1/4 in QD cores, respectively, while the gross amount of Zn and In precursors and all other variables were remained constant. Cu+ ion doping concentration in both samples was fixed at 5 mol%. The Mn:ZnInS/ZnS core/shell QDs was achieved by hot injection method as described in ref. 9. The Mn:ZnInS/ZnS core/shell QDs (λ = 600 nm, PL QY = 56%) was prepared with Zn/In precursor ratio of 1/1 in QD cores and Mn2+ ion doping concentration of 4.5 mol%. The CdSe/ZnS core/shell QDs with PL QY of 90% were synthesized by SILAR method as described in ref. 19.The QD films were obtained by drop-casting QDs chloroform solution onto silicon substrates. As reference samples, the QD/PMMA blend films were made by drop-casting from the QDs solution blending with PMMA (poly(methyl methacrylate), Mw = 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) onto silicon substrates. The molar ratio of PMMA to QD was about 105.
000) onto silicon substrates. The molar ratio of PMMA to QD was about 105.
2.2 Characterization
Structure, morphology and sizes of the obtained QDs were characterized by the X-ray diffraction (XRD, Rigaku D/max-2500) and transmission electron microscopy (TEM, JEM-2100HR). UV-visible measurements were carried out on UV-Vis-NIR scanning spectrophotometer (UV-5800 PC). Steady-state and time-resolved PL spectra were measured by spectrometer (Horiba Jobin Yvon Fluorolog-3) with a QY accessory and a time-correlated single-photon counting spectrometer (TCSPC). The continuous excitation source was a 150 W ozone-free xenon arc-lamp. The PL decay curves of Cu or Mn doped QDs were fitted by: I(t) = A1exp(−t/τ1) + A2exp(−t/τ2) and the average lifetime τAV were calculated by τAV = (A1τ12 + A2τ22)/(A1τ1 + A2τ2), where A1 and A2 are fractional contributions of PL decay lifetimes τ1 and τ2. While the PL decay curves of CdSe/ZnS QDs were fitted well using a single exponential function.
3. Results and discussion
Fig. 1 shows the UV-visible absorption and PL spectra for Cu doped Zn–In–S/ZnS QDs with Zn/In precursor ratios of 1/2 and 1/4 and Mn:Zn–In–S/ZnS QDs with Zn/In precursor ratio of 1/1 in chloroform solution. From Fig. 1(a) and (b), it can be found that both the absorption onset and PL peak of Cu-doped QDs can be tunable through changing Zn/In molar ratio in the QD cores. There is general consensus that the PL of Cu doped QDs originates from the recombination of the electron from the conduction band of host with the hole in the Cu T2 state above the valence band.7,12,20 Thus, the Cu-doped QDs could exhibit a color tunable emission by changing the composition of Zn–In–S alloyed host due to the bandgap of ZnS (3.7 eV) and In2S3 (2.1 eV). The full widths at half-maximum (FWHM) of the PL bands are estimated to about 100 and 125 nm for Cu:Zn–In–S/ZnS QDs centered at 580 nm (called as Cu-QD580) and 620 nm (called as Cu-QD620), respectively, as listed in Table 1. The wide FWHM for Cu doped QDs is expected to come from the different distributions of sizes or copper energy levels.21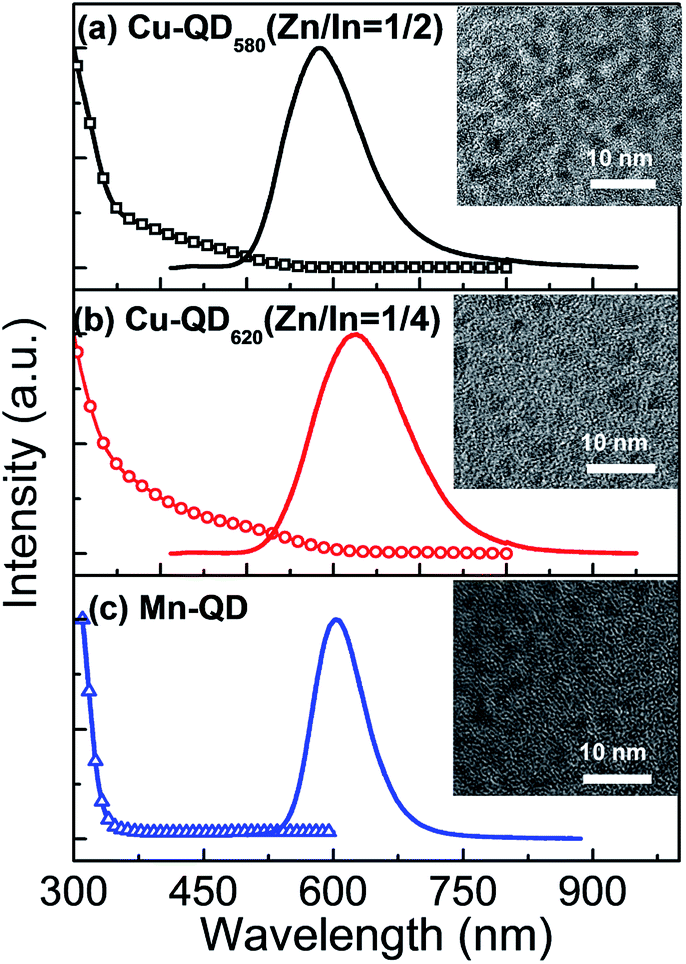 | ||
| Fig. 1 UV-visible absorption (dashed lines) and normalized PL spectra (solid lines) of Cu:Zn–In–S/ZnS core/shell QDs with Zn/In ratios of 1/2 (a) and 1/4 (b), and Mn:Zn–In–S/ZnS core/shell QDs (c) in chloroform. The insets display the corresponding TEM images of QDs. | ||
| Sample | PL peak (nm) | Stokes shift (nm) | FWHM (nm) | PL QYs |
|---|---|---|---|---|
| Cu-QD580 | 580 | 100 | 100 | 0.47 |
| Cu-QD620 | 620 | 160 | 125 | 0.40 |
| Mn-QD | 600 | 270 | 70 | 0.56 |
In Fig. 1(c), the PL emission of the Mn:Zn–In–S/ZnS QDs is located at 600 nm (called as Mn-QD) with an FWHM of 70 nm, which belongs to the typical emission of Mn2+ ions from 4T1–6A1 internal d–d transition in Zn–In–S host.9 It has been previously reported that the emission from Mn2+ ions doped in II–VI host nanocrystals (e.g. ZnS, ZnSe and CdS) is usually centered at about 580 nm.22,23 The difference in PL emission energy could be ascribed to Mn2+ ions in different radical positions of host nanocrystals because the emission of Mn2+ ions is very sensitive to the symmetry of the crystalline field of host. Compared with Cu doped QDs, a larger Stokes shift of about 270 nm for Mn doped QDs is observed, which can be attributed to the significant difference in PL mechanism. Representative TEM images for Cu or Mn doped QDs are shown in the inset of Fig. 1. It can be seen that all three QDs samples are monodispersed with a near-spherical shape and the average sizes of 4.3, 4.6, and 3.4 nm are estimated for Cu-QD580, Cu-QD620, and Mn-QD, respectively.
Fig. 2 displays the steady-state and time-resolved PL spectra of Cu or Mn doped QDs in solution and films. The PL peaks are observed at 587 and 651 nm for Cu-QD580 and Cu-QD620 films, respectively. They all exhibit a red-shifted PL peak compared to the corresponding QDs in solution. In addition, the PL decay curves were well fitted by bi-exponential function with time constants of 50–130 ns and 220–340 ns and the fitting results are summarized in Table 2. The average PL lifetimes decrease from 267.8 and 232.2 ns to 224.4 and 182.2 ns for Cu-QD580 and Cu-QD620, respectively, when the QDs are in solution and films. The obvious decrease in PL lifetime is also observed for Mn doped QD films, although the red shift in PL peak is not found, as shown in Fig. 2(c) and (f).
 | ||
| Fig. 2 PL spectra (top) and decay curves (bottom) of Cu or Mn doped Zn–In–S/ZnS QDs in solution (black line), films (red line) and QD/PMMA blend films (green line). | ||
| Sample | τ1 | A1 | τ2 | A2 | τAV | |
|---|---|---|---|---|---|---|
| Cu-QD580 | Solution | 332.5 ns | 0.45 | 125.8 ns | 0.54 | 267.8 ns |
| Pure film | 282.4 ns | 0.42 | 102.4 ns | 0.55 | 224.4 ns | |
| PMMA film | 278.2 ns | 0.46 | 95.0 ns | 0.53 | 226.5 ns | |
| Cu-QD620 | Solution | 253.1 ns | 0.62 | 58.2 ns | 0.33 | 232.2 ns |
| Pure film | 221.4 ns | 0.48 | 83.5 ns | 0.50 | 182.5 ns | |
| PMMA film | 243.6 ns | 0.40 | 98.7 ns | 0.59 | 189.4 ns | |
| Mn-QD | Solution | 6.0 ms | 0.73 | 1.3 ms | 0.26 | 5.6 ms |
| Pure film | 3.8 ms | 0.64 | 1.2 ms | 0.35 | 3.5 ms | |
| PMMA film | 4.4 ms | 0.54 | 1.7 ms | 0.45 | 3.7 ms | |
Previous works have been reported that the differences in the PL peaks and decay lifetimes between the QDs in solution and films can be attributed to the energy transfer from small QDs to large ones due to the size distribution.24,25 Thus, it is important to confirm whether the energy transfer occurs in Cu or Mn doped QD films. According to the Förster theory, the energy transfer efficiency is dependent on the spectral overlap between the emission and absorption spectra of QDs.26 However, it can be seen from Fig. 1 that the spectral overlap between the absorption and emission spectra of Cu or Mn doped QDs is expected to be so small that the energy transfer between Cu (or Mn):ZnInS/ZnS QDs can be negligible.
On the other hand, we prepared the QD/PMMA blend films, in which PMMA can sufficiently insulate QDs to avoid Förster energy transfer between differently sized QDs due to their aggregation. The PL spectra and decay curves of QD/PMMA blend films are also shown in Fig. 2, the corresponding fitting time constants are also summarized in Table 2. It can be found that both the PL peak and lifetime of Cu or Mn doped QD films with and without PMMA are almost the same, which further reveals that no energy transfer process takes place in the Cu or Mn doped QD films because the energy transfer process would result in a decrease in PL lifetime because it is an additional nonradiative de-excitation path for excited electrons.26 Moreover, the PMMA in blend films also can reduce or avoid the possible charge transfer between QDs with different sizes.
Because II–VI CdSe/ZnS QDs are commonly used to fabricate optoelectronic devices, we will study the PL properties of CdSe/ZnS QD films. Fig. 3 shows the PL spectra and decay curves of CdSe/ZnS QDs in solution and films. In comparison, the PL peak shifts from 623 to 633 nm and the average PL lifetime decreases from 25.4 to 12.8 ns when the CdSe/ZnS QDs are in solution and films. However, the PL peak blue-shifts to 628 nm and the lifetime increases to 18.7 ns upon blending with PMMA. Since the energy transfer was tentatively restrained in QD/PMMA blending films due to the isolated QDs, the observation of these phenomena provides a proof to confirm the existence of QD-to-QD energy transfer in CdSe/ZnS QD films. It is worth noting that compared with that of QDs solution, there are only about 16.2%, 21.5%, and 37.5% loss of PL lifetimes for the Cu-QD580, Cu-QD620, and Mn-QD films, respectively, whereas the reduction in PL lifetime of CdSe QDs reaches about 50%. The less reduction in PL lifetimes of Cu or Mn doped QD films can be ascribed to negligible energy transfer between QDs.
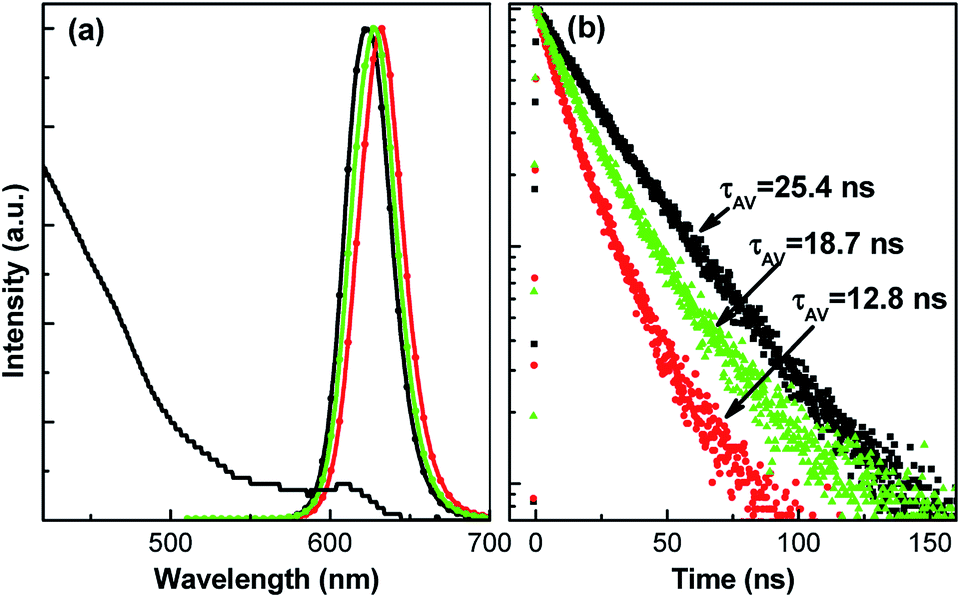 | ||
| Fig. 3 UV-visible absorption and PL spectra (a) and decay curves (b) of CdSe/ZnS QDs in chloroform (black line), films (red line) and QD/PMMA blend films (green line). | ||
Although no energy transfer takes place in Cu or Mn doped QD films, the red-shifted emission peak and decreased lifetime are still found in contrast to that of the corresponding QDs solution, indicating that there are other factors to influence the PL properties of QD films. Previous studies have suggested that several surface defects, such as vacancies, and dangling bonds, could act as trapping sites for photo-excited carriers, which decreases the PL lifetime by an accompanying increase of the non-radiative decay.27,28 Thereby, we postulate that the reduced lifetimes observed in doped QD films are mainly considered to result from the formation of surface traps/defects due to the loss of surface ligands when the QDs are deposited from solution to film. In order to get a deeper insight into the PL properties of Cu or Mn doped Zn–In–S QD films, the PL decays of the QDs solution and pure QD films were measured at various detection wavelength around PL emission peak at an interval of 15 or 10 nm, as shown in Fig. 4. As reference, the PL decay curves of CdSe/ZnS QDs in solution and PMMA films are also shown in Fig. 4(d) and (h). Fig. 5 summarizes the dependence of average PL lifetimes on the detected wavelength.
 | ||
| Fig. 4 PL decay curves of the QDs in solution (top) and solid films (bottom) detected at different wavelength around PL emission peak: Cu-QD580 (a and e), Cu-QD620 (b and f), Mn-QD (c and g). As reference, the PL decay curves of CdSe/ZnS QDs in solution and PMMA film are also shown in (d and h). | ||
 | ||
| Fig. 5 The dependence of PL lifetimes of QDs in solution (black triangles) and PMMA films (red circles) on the detected wavelengths. | ||
As seen in Fig. 5(a), the average lifetime of Cu-QD580 in solution almost keeps at about 280 ns when the detected emission wavelength increases from 550 nm to 610 nm, which may mean that the PL emission of the Cu-QD580 mainly comes from the recombination of the electron at conduction band with Cu-related acceptors. However, the decay time of the QDs in solid films is slightly dependent on the emission energy. The lifetime increases gradually from 205 to 238 ns as the detected emission wavelength increases from 557 to 617 nm. Such an emission-wavelength-dependent behavior of the PL lifetime is characteristic property of donor–acceptor pair (DAP) recombination.29 The various PL lifetimes of Cu-QD580 in solid films reveal that apart from the recombination between conduction band and acceptor, DAP recombination also exists in the Cu-QD580 films. In addition, when QDs was deposited as solid films, some new defects/traps due to the loss of surface ligands also led to the formation of nonradiative recombination centers, thereby, reducing the PL lifetime. Similar results were also observed CdSe/ZnS QDs in solution and PMMA films.
It is noted that the PL lifetimes for Mn doped QDs in solution and films are all independent on the emission wavelength because Mn dopant emission comes from the intrinsic 4T1–6A1 transition of Mn2+ ions via energy transfer from the host nanocrystals30–32 and new defects/traps on the surface of QDs only can increase the numbers of nonradiative recombination centers and reduce PL lifetime. For Cu-QD620, it can be seen from Fig. 5(b) that the PL lifetime of Cu-QD620 in solution significantly increases with the detected emission wavelength, which indicates that two radiative recombination processes, including the recombination between conduction band to acceptors and DAP recombination, are contributed to the PL emission. But, the PL lifetime of Cu-QD620 films showing a fast increase with detected wavelength also can reveal the formation of new defects/traps on the surface of QDs. On the other hand, compared with that of QDs in solution, the red-shifted PL peak observed in Cu or Mn doped QD films can be explained by the significant PL quenching of smaller QDs due to the size distribution of QDs, because small QDs have more numerous defects on their surface, which would reduce the stability of QDs in solid films.
In our work, the Cu:Zn–In–S/ZnS QDs with a thick shell were also synthesized through increasing Zn precursor solution during shell growth process. Fig. 6 shows the PL spectra and decay curves of Cu-QD620 with a thick shell in solution and films. The size of Cu-QD620 increases to 6.8 nm estimated by TEM image (shown in the inset of Fig. 6(a)) and the PL quantum yield also increases from 40% to 50% due to the increase of shell thickness. It is exciting to see that the red-shift in PL peak is only 10 nm after the QD solid films was formed, accompanied by 6.7% shortening of PL lifetime. The reduction in PL lifetime of Cu-QD620 with a thick shell only reaches 6.7%, while that is 21.5% for Cu-QD620 with a thin shell. This indicates that increasing the shell thickness can significantly restrain the roll-off of luminous efficiency of Cu or Mn doped QDs in films because the surface nonradiative recombination centers are significantly passivated by a thick shell.33
 | ||
| Fig. 6 PL spectra (a) and decay curves (b) of Cu-QD620 with a thick shell in chloroform (black line) and films (red line). The inset displays the corresponding TEM image of QDs. | ||
4. Conclusions
In summary, we have studied the PL properties of Cu or Mn doped Zn–In–S/ZnS core/shell QDs in solution and solid films. Both the PL peaks and lifetimes almost exhibited no variation for Cu or Mn doped Zn–In–S/ZnS QD solid films compared with that of the corresponding QDs/PMMA films, indicating that no energy transfer process occurred in the doped QD films. An obvious energy transfer was found in the CdSe/ZnS QD films. The considerable difference was attributed to the different PL mechanism of two kinds of QDs. Compared with QDs solution, the PL lifetimes of Cu or Mn doped QD films only dropped 16.2–37.5% because of no energy transfer between QDs, whereas that of CdSe/ZnS QD films decreased by 50%. The decrease in PL lifetime found in Cu or Mn doped QD films could be attributed to the formation of surface traps/defects due to the loss of surface ligands when QDs were deposited as solid films. Moreover, the small change in PL lifetime could be observed in QD films by increasing the shell thickness of doped QDs. The experimental results are beneficial to the design of photoelectronic devices based on Mn and Cu doped Zn–In–S/ZnS QDs.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21371071 and 11274304) and Key Program for the Development of Science and Technology of Jilin Province (No. 20150204067GX).Notes and references
- O. Chen, J. Zhao, V. P. Chauhan, J. Cui, C. Wong, D. K. Harris, H. Wei, H.-S. Han, D. Fukumura, R. K. Jain and M. G. Bawendi, Nat. Mater., 2013, 12, 445–451 CrossRef CAS PubMed.
- W. T. Yang, W. S. Guo, X. Q. Gong, B. B. Zhang, S. Wang, N. Chen, W. T. Yang, Y. Tu, X. M. Fang and J. Chang, ACS Appl. Mater. Interfaces, 2015, 7, 18759–18768 CAS.
- M. Smith, H. W. Duan, A. M. Mohs and S. M. Nie, Adv. Drug Delivery Rev., 2008, 60, 1226–1240 CrossRef PubMed.
- E. H. Sargent, Nat. Photonics, 2012, 6, 133–135 CrossRef CAS.
- W. Y. Ji, P. T. Jing, W. Xu, X. Yuan, Y. J. Wang, J. L. Zhao and A. K.-Y. Jen, Appl. Phys. Lett., 2013, 103, 053106 CrossRef.
- H. L. Huang, F. C. Zhao, L. G. Liu, F. Zhang, X. G. Wu, L. J. Shi, B. S. Zou, Q. B. Pei and H. Z. Zhong, ACS Appl. Mater. Interfaces, 2015, 7, 28128–28133 Search PubMed.
- W. J. Zhang, Q. Lou, W. Y. Ji, J. L. Zhao and X. H. Zhong, Chem. Mater., 2014, 26, 1204–1212 CrossRef CAS.
- T. T. Jiang, J. L. Q. Song, H. J. Wang, X. C. Ye, H. Wang, W. T. Zhang, M. Y. Yang, R. X. Xia, L. X. Zhu and X. L. Xu, J. Mater. Chem. B, 2015, 3, 2402–2410 RSC.
- S. Cao, J. L. Zhao, W. Y. Yang, C. M. Li and J. J. Zheng, J. Mater. Chem. C, 2015, 3, 8844–8851 RSC.
- S. Cao, C. M. Li, L. Wang, M. H. Shang, G. D. Wei, J. J. Zheng and W. Y. Yang, Sci. Rep., 2014, 4, 7510–7518 CrossRef PubMed.
- P. Zhou, X. S. Zhang, L. Li, X. J. Liu, L. L. Yuan and X. G. Zhang, Opt. Mater. Express, 2015, 5, 2069–2080 CrossRef.
- X. S. Tang, Z. Z. Zu, L. F. Bian, J. H. Du, W. W. Chen, X. F. Zeng, M. Q. Wen, Z. G. Zang and J. M. Xue, Mater. Des., 2016, 91, 256 CrossRef CAS.
- S. C. Erwin, L. Zu, M. I. Haftel, A. L. Efros, T. A. Kennedy and D. J. Norris, Nature, 2005, 436, 91–94 CrossRef CAS PubMed.
- X. Yuan, J. Hua, R. S. Zeng, D. H. Zhu, W. Y. Ji, P. T. Jing, X. D. Meng, J. L. Zhao and H. B. Li, Nanotechnol., 2014, 25, 435202 CrossRef PubMed.
- S. Sarkar, N. S. Karan and N. Pradhan, Angew. Chem., Int. Ed., 2011, 50, 6065–6069 CrossRef CAS PubMed.
- B. K. Chen, H. Z. Zhong, M. X. Wang, R. B. Liu and B. S. Zou, Nanoscale, 2013, 5, 3514–3519 RSC.
- Y. Liu, X. Zhang, Z. Hao, Y. Luo, X. Wang and J. Zhang, J. Mater. Chem., 2011, 21, 16379–16384 RSC.
- Y. Liu, X. Zhang, Z. Hao, X. Wang and J. Zhang, J. Mater. Chem., 2011, 21, 6354–6358 RSC.
- R. G. Xie, U. Kolb, J. Li, T. Basche and A. Mews, J. Am. Chem. Soc., 2005, 127, 7480–7488 CrossRef CAS PubMed.
- K. E. Knowles, H. D. Nelson, T. B. Kilburn and D. R. Gamelin, J. Am. Chem. Soc., 2015, 137, 13138–13147 CrossRef CAS PubMed.
- S. Brovelli, C. Galland, R. Viswanatha and V. I. Klimov, Nano Lett., 2012, 12, 4372–4379 CrossRef CAS PubMed.
- S. Niladri Karan, D. D. Sarma, R. M. Kadam and N. Pradhan, J. Phys. Chem. Lett., 2010, 1, 2863–2866 CrossRef.
- W. Xu, X. D. Meng, W. Y. Ji, P. T. Jing, J. J. Zheng, X. Y. Liu, J. L. Zhao and H. B. Li, Chem. Phys. Lett., 2012, 532, 72–76 CrossRef CAS.
- N. Droseros, K. Seintis, M. Fakis, S. Gardelis and A. G. Nassiopoulou, J. Lumin., 2015, 167, 333–338 CrossRef CAS.
- K. Furuta, M. Fujii, H. Sugimoto and K. Imakita, J. Phys. Chem. Lett., 2015, 6, 2761–2766 CrossRef CAS PubMed.
- J. R. Lakowicz, Principles of fluorescence spectroscopy, 3rd edn, Springer-Verlag, Berlin, Germany, 2006, pp. 443–527 Search PubMed.
- H. Lifshitz, I. Dag, I. D. Litvin and G. Hodes, J. Phys. Chem. B, 1998, 102, 9245–9250 CrossRef.
- D. F. Underwood, T. Kippeny and S. J. Rosenthal, J. Phys. Chem. B, 2001, 105, 436–443 CrossRef CAS.
- Y. Hamanaka, T. Kuzuya, T. Sofue, T. Kino, K. Ito and K. Sumiyama, Chem. Phys. Lett., 2008, 466, 176–180 CrossRef CAS.
- G. Manna, S. Jana, R. Bose and N. Pradhan, J. Phys. Chem. Lett., 2012, 3, 2528–2534 CrossRef CAS PubMed.
- H. Y. Chen, T. Y. Chen and D. H. Son, J. Phys. Chem. C, 2010, 114, 4418–4423 CAS.
- T. Kuroda, H. Ito, F. Minami and H. Akinaga, J. Lumin., 1997, 72, 106–107 CrossRef.
- X. Yuan, J. J. Zheng, R. S. Zeng, P. T. Jing, W. Y. Ji, J. L. Zhao, W. Y. Yang and H. B. Li, Nanoscale, 2014, 6, 300–307 RSC.
| This journal is © The Royal Society of Chemistry 2016 |