
Chemical diversification of essential oils, evaluation of complex mixtures and identification of a xanthine oxidase inhibitor†
P. Garcíab,
I. A. Ramallob,
M. O. Salazarab and
R. L. E. Furlan*ab
aFarmacognosia, Facultad de Ciencias Bioquímicas y Farmacéuticas, Universidad Nacional de Rosario, 2000, Suipacha 531, Rosario, Argentina
bInstituto de Investigaciones para el Descubrimiento de Fármacos de Rosario (IIDEFAR, CONICET-UNR), Ocampo y Esmeralda, 2000, Rosario, Argentina. E-mail: furlan@iidefarconicet.gob.ar
First published on 6th June 2016
Abstract
A set of chemically engineered essential oils has been generated through chemical diversification by reaction with bromine. The impact of the reaction over the chemical composition of the mixtures was qualitatively demonstrated through GC-MS and utilizing multivariate analysis of 1H NMR and GC-MS. Most of the components of the essential oils are transformed by the reaction expanding the chemical diversity of the mixtures. Biological changes between essential oils and brominated essential oils were demonstrated through image analysis of xanthine oxidase autography profiles. The highest biological activity increase was obtained for the Foeniculum vulgare Mill essential oil. Coupling of xanthine oxidase autography with the BIOMSID strategy allowed the identification of the molecular formula of the active compound. Bioguided fractionation of the mixture led to the isolation of (RS)-2-bromo-1-(4-methoxyphenyl) propan-1-one for being responsible for the observed bioactivity. This xanthine oxidase inhibitor could have been formed from the inactive natural component anethole. The inhibitory potency of this semisynthetic compound was in the same order of magnitude as allopurinol, the most used inhibitor.
Introduction
Because of biological selection, natural products (NPs) are an excellent source of substructures for the design of new drugs.1 Taking advantage of the biomolecular recognition properties of natural products, different strategies have been developed that allow the exploration of potentially useful scaffolds inspired by Nature.2–5One such strategy is the chemical diversification of natural product mixtures to produce chemically engineered extracts (CEEs).6–11 This approach involves the chemical transformation of reactive fragments commonly found in natural products, to introduce elements or functionalities that are relevant for bioactivity.12 Its application to different natural extracts has led to the discovery of several bioactive compounds.6–11,13–16
Halogenation reactions are important medicinal chemistry tools to alter biological properties of molecules,17,18 however natural organohalogen compounds are very uncommon.19
Although some antibacterial,20 antitumor,21 antiviral22 and antifungal23 brominated metabolites have been isolated from corals, shellfish, algae and marine sponges, terrestrial plants rarely produce bromine containing compounds.24 In a previous report, a brominated psolarene, inhibitor of the enzyme acetylcholinesterase, was isolated from a chemically engineered extract of Conium maculatum L.13
Within the natural product extracts, the essential oils (EOs) are multi-component systems25 composed of low-molecular weight lipophilic compounds derived from different biosynthetic pathways.26 In general, its production by plants is diversity oriented, with the resulting generation of complex mixtures of compounds. This strategy suggests a broadly-tuned defense system that has the potential to regulate not only plant–insect, but also plant–mammal interactions. Consequently, the bioactive volatilome is now emerging as a novel potential source of interesting lead structures for drug discovery.26
Xanthine oxidase (XO) is an enzyme from the purine salvage pathway that catalyses the oxidation of hypoxanthine to xanthine with subsequent production of uric acid and reactive oxygen species from xanthine oxidation. This enzyme is an important target for various therapeutic indications such us ischemia-reperfusion injury, gout and tumor lysis syndrome, circulatory shock, chronic heart failure and vascular and inflammatory diseases.27 Despite the effectiveness of known purine based xanthine oxidase inhibitors, some serious side effects of these drugs has revived the interest for the search of new non-purine XO inhibitors.28–33
Here we report the chemical diversification of a series of EOs through bromination to produce mixtures with completely different composition and biological properties. Combination of a variety of analytical tools that involve the use of chromatography, spectrometry, multivariate analysis and bioassays allowed the fast: (a) generation of a high number of natural products derivatives, (b) evaluation of its chemical and biomolecular properties without purification, (c) identification of a XO inhibitor and (d) its straightforward bioguided isolation.
Results and discussion
Double bound as target
The success of the CEE approach relies upon the power of numbers: in order to increase the chances of generating a bioactive compound, it is crucial to produce many compounds that incorporate the desired chemical feature or element. This can be achieved finding fragments that are: (a) very common within the components of the starting mixtures and (b) reactive towards the reaction conditions.Essential oils include in their composition aromatic and more or less saturated aliphatic compounds, with different degree of oxidation. 78% of the EO constituents present in the Dictionary of Natural Products (DNP) (Chapman & Hall/CRC, 2001) include at least one double bound in their structures, whereas 22% contain at least one aromatic double bond.
On the contrary, halogenated NPs are marginal within the molecules present in the DNP database. For example, only 1.4% are organobromine compounds, and none of these halogenated molecules are EO constituents (see ESI† for searching strategy in DNP). In this context, the introduction of bromine using double bonds and aromatic rings as entry points could be a promising strategy to produce unnatural derivatives of natural products from EOs.
Aiming at the diversification of the components of EOs through chemical alteration of these functionalities, we tested the bromination reaction on 17 EOs from plants belonging to different genus. The reaction was carried out following the protocol reported by Méndez et al.13 with minor changes, giving 17 brominated essential oils (BEOs).
Chemical impact evaluation
In all cases the reaction produced a total-mass increase that ranged from 121%, for the EO of Melaleuca viridiflora Sol. ex Gaertn., to more than 145% for the EO of Cymbopogon martinii (Roxb.) W. Watson. This increase could result from the incorporation of bromine in some of the EO components.To gain insight into the chemical differences between the starting EOs and the BEOs, all the mixtures were evaluated employing GC-MS and 1H NMR. GC-MS analysis indicates that the BEOs include higher numbers of compounds than the EOs. Also, most of the constituents of the BEOs are different from the constituents of the starting EOs and contain bromine in their structure. On average, 87% of the peaks detected in the EO chromatograms disappeared because of the reaction, and 95% of the peaks detected in the BEO chromatograms were absent in the parent EOs (Fig. 1a). 81% of these new peaks, showed the characteristic isotopic pattern of organobromine compounds whereas the 19% left of new peaks could be oxidation products.
 | ||
| Fig. 1 Box and whiskers plot for (a) percentage of peaks that disappeared from the chromatograms because of the reaction (blue), percentage of peaks that appeared in the chromatograms after the reaction (red) and percentage of brominated peaks that appeared in the chromatograms after the reaction (orange), (b) number of peaks detected in the EOs (blue) and BEOs (red) GC-MS chromatograms. | ||
Additionally, the average number of detected compounds in BEOs was 2.2 times higher than in EOs, suggesting that around 2 products were produced from each natural molecule present in the starting mixture (Fig. 1b).
GC-MS data analysis also shows that the compounds present in the group of BEOs cover a broader range of retention times, and their ionized fragments cover a broader range of m/z values (Fig. 2). Since both ranges are expanded towards higher values, this expansion could be indicative of an increase in molecular weight and a decrease in polarity of the molecule population, as could be expected after bromination.
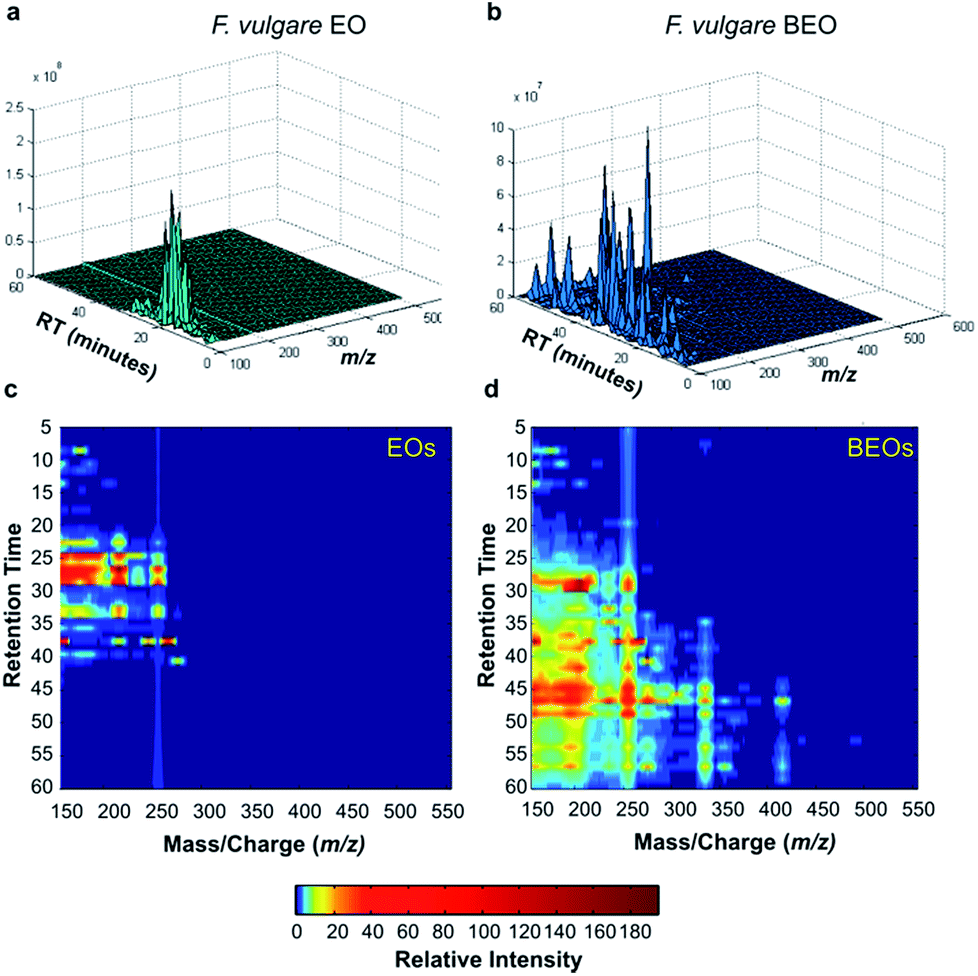 | ||
| Fig. 2 (a) GC-MS data of F. vulgare EO. (b) GC-MS data of F. vulgare BEO. (c) Colour coded relative intensity of chromatographic peaks (retention time) and MS signals, present in the GC-MS data obtained from the 17 EOs. (d) Colour coded relative intensity of chromatographic peaks and MS signals, present in the GC-MS data obtained from the 17 BEOs. | ||
The GC-MS data were also analysed with the non-supervised method Principal Component Analysis (PCA). The score plot of PC1 and PC2 showed separation of the samples mainly along the second principal component (Fig. 3a).
 | ||
| Fig. 3 Principal component analysis. (a) Score plot of the GC-MS data. BEOs (red) and EOs (blue). (b) Loading plot of the PC2. | ||
According to PC2 loading plot, buckets with m/z values below 200, and retention times between 5 and 10 minutes or between 20 and 50 minutes have the largest contribution in the separation observed in the score plot (Fig. 3b): red buckets, relatively more important in BEOs than in EOS, contribute with a negative effect in PC2, whereas the blue buckets, relatively more important in EOs, contribute with a positive effect in PC2.
These changes in composition were also evident from 1H NMR analysis coupled to PCA. Again, EOs and BEOs could be differentiated mainly along PC2 (Fig. 4a). In this case, three regions involved in the discrimination of the BEOs from EOs were identified in the PC2 loading plot. The most influential region was between 4.9 ppm and 5.7 ppm, and corresponds to double bond 1H signals (one of the target groups for bromination) (Fig. 4b). Another important region was comprised between 5.8 and 6.1 ppm and corresponds to double bond hydrogen signals conjugated with aromatic rings. Finally, a third region was detected between 1.6 and 1.7 ppm that corresponds to methylene 1H signals that are in α position to double bonds (Fig. 4b). It is clear that the functional group most affected by the reaction is the double bond, so compounds containing double bonds are present in EOs and are marginal in BEOs.
 | ||
| Fig. 4 Principal component analysis. (a) Score plot of the 1H NMR data. BEOs (red) and EOs (blue). (b) Loading plot of the PC2. | ||
Without knowing the exact composition of any of the 34 mixtures, altogether these analytical data indicate that most of the components in the starting mixtures were altered, mainly by chemical transformation of their double bonds to introduce one or more bromine atoms, producing on average more than one derivative from each natural precursor.
Biological impact evaluation
The effect of the reaction on the biomolecular properties of the mixtures was initially evaluated by measuring XO inhibition by thin layer chromatography (TLC) autography, an appropriate technique for bioactivity analysis of complex mixtures.34 In XO TLC-autography the samples are developed onto a TLC plate which is then covered with a gel containing XO and a revealing agent. Immersion of the gel layer-plate in substrate solution makes evident the presence of an inhibitor through the colourless halo it produces.35 Comparison of the bioactivity profile for each pair of EO/BEO was performed using 1-D gels analysis. The XO inhibition profile (Fig. 5b) obtained for each EO was subtracted from the inhibition profile of its derived BEO (Fig. 5a). The area under the resulting curve shows the change on XO-inhibitory activity produced by the reaction for each EO/BEO couple (Fig. 5c). | ||
| Fig. 5 Typical processing of the autographies. (a) BEO profile. (b) EO profile. (c) EO/BEO profile subtraction. (d) Changes observed in XO inhibition by the bromination reaction over EOs. Blue bars illustrate de generation of biological activity and red bars indicate the biological activity that was only present in the EOs and that disappears after reaction. By meaning of abbreviations see material section. | ||
Positive blue bars shown in Fig. 5d indicate that biological activity was generated by the reaction whereas negative red bars indicate that inhibition was decreased after bromination. The highest XO inhibitory activity generation was observed for the essential oil obtained from Foeniculum vulgare Mill. (‘FV’ in Fig. 5d). The TLC of F. vulgare EO/BEO revealed with UV (365 and 254 nm) with a chemical reagent and with the XO assay is shown in Fig. 6.
 | ||
Fig. 6 TLC of F. vulgare EO and F. vulgare BEO revealed with: (a) p-anisaldehyde-sulphuric acid. (b) UV 365 nm. (c) UV 254 nm. (d) Vainillin-sulphuric acid. (e) XO assay. Mobile phase hexane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethyl acetate (90:10). :ethyl acetate (90:10). | ||
BIOMSID analysis and bioassay-guided fractionation
To gain insight into the identity of the compound responsible of the observed activity in the F. vulgare BEO, the BIOMSID (BIOautography coupled to Mass Spectrometry for the IDentification of compounds)36 strategy was applied.BIOMSID allows coupling TLC-autography data with High Resolution Mass Spectrometry (HRMS) in order to link the bioactivity observed in a complex mixture with the molecular formula of the compound that is responsible for such bioactivity. Although this strategy has only been used with an acetylcholinesterase assay to dereplicate natural extracts,36 in principle it could be applied to other enzymes, such as XO, and to other bioactive complex mixtures, such as the BEOs.
To implement BIOMSID, F. vulgare BEO samples were chromatographed on TLC using three different conditions and the plates were tested for XO activity using the autographic assay. Samples from the inhibition zones and from the background were taken from each TLC plate and subjected to HRMS analysis. The obtained spectra were processed with a MATLAB algorithm that compares the spectra searching for common signals that can be linked to the structure of the active compound.
Four signals were linked to the possible bioactive structure with m/z values 243.0015, 244.9995, 264.9835 and 266.9815, which may correspond to the [M + H]+, [(M + 2) + H]+, [M + Na]+, and [(M + 2) + Na]+ ions respectively (Fig. 7). These values fit with the characteristic isotopic pattern of brominated compounds.37 Although these signals, corresponding to the active compound, were picked out from a crowded zone of the spectra (Fig. 7a and b), they could be correctly assigned, and verified for the C10H11BrO2 molecular formula.
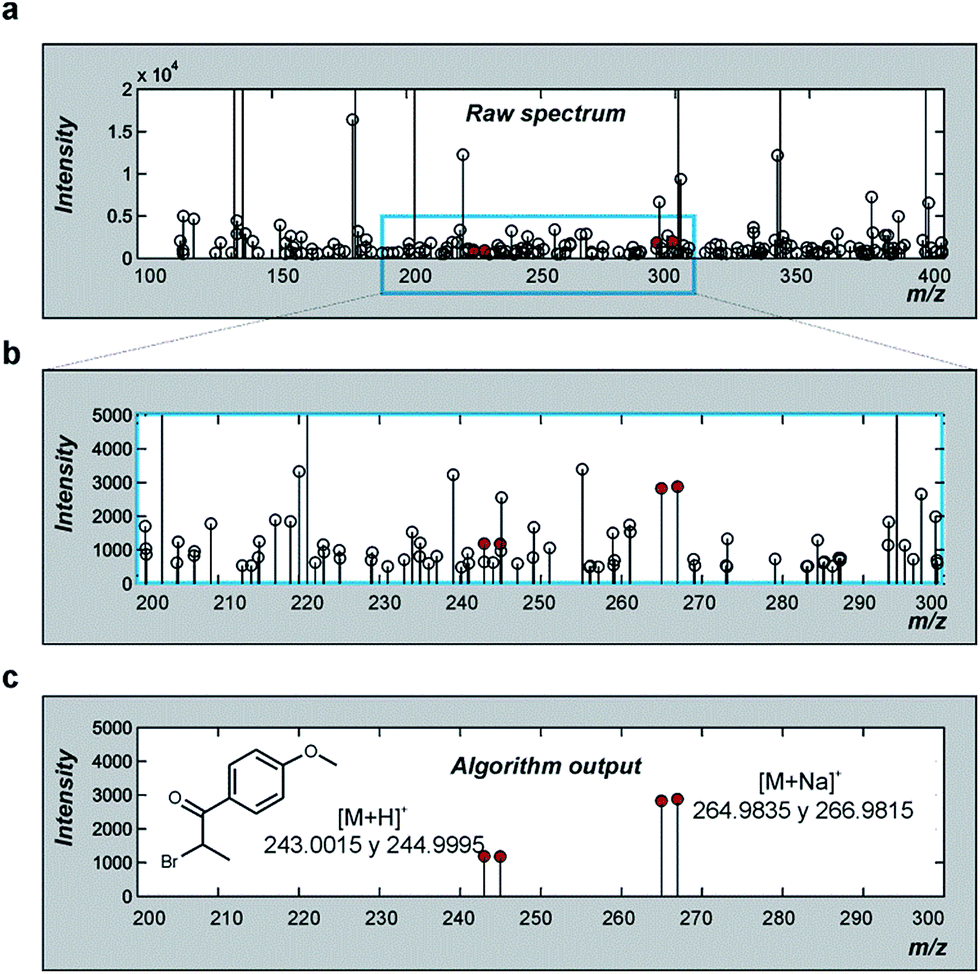 | ||
| Fig. 7 BIOMSID analysis of the F. vulgare BEO mass spectra. (a) Raw data from TLC-autography inhibition spot. (b) Region of the raw spectrum where the selected signals are located (c) algorithm output: signals proposed for the active compound. | ||
Bioassay-guided fractionation of the F. vulgare BEO led to the isolation of (RS)-2-bromo-1-(4-methoxyphenyl) propan-1-one (1, Scheme 1) as responsible for the observed XO inhibition activity. The HRMS spectrum of 1 showed m/z values that were equal to those previously identified by BIOMSID.
 | ||
| Scheme 1 Synthesis of (RS)-2-bromo-1-(4-methoxyphenyl) propan-1-one (1) from anethol (2) with bromine. | ||
This XO inhibitor could have been formed from the inactive natural component anethole (2, Scheme 1) whose presence in F. vulgare EO was confirmed by GC-MS. The origin of 1 was demonstrated treating anethole with bromine under the same conditions previously used for the bromination of the EOs and obtaining the compound 1 in 3.9% yield (Scheme 1).
The inhibitory potency of 1 was comparable to the reference XO inhibitor allopurinol.38 The observed IC50 for 1 was 8.23 μM, whereas an IC50 = 2.62 μM was observed for the reference inhibitor. It is worth mentioning that under these experimental conditions the IC50 observed for the natural precursor anethole was higher than 200 μM.
Materials and methods
Materials
XO (EC 1.1.3.22; product no. X-1875) was purchased from Sigma (St Louis, MO, USA). Xanthine was purchased from Fluka (Buchs, Switzerland). Ethylenediaminetetraacetic acid (EDTA), and nitroblue tetrazolium (NBT) were purchased from Aldrich (Milwaukee, WI, USA). Aluminum-blacked silica gel 60 F254 TLC layers were purchased from Merck (Darmstadt, Germany). Agar was purchased from Britania (Buenos Aires, Argentina).The EOs Abies alba Mill. (‘AAL’), Citrus aurantifolia (Christm.) Swingle (‘CAF’), Cinnamomum cassia (Nees & T. Nees) J. Presl (‘CC’), Citrus limonum (L.) Burm. f. (‘CL’), Cymbopogom martinii (Roxb.) W. Watson (‘CM’), Canagna odorata (Lam.) Hook. f. & Thomson (‘CO’), Coriandrum sativum L. (‘COS’), Cinnamomum verum J. Presl (‘CV’), Cymbopogom citratus (DC.) Stapf (‘CYC’), Foeniculum vulgare Mill. (‘FV’), Juniperus communis L. (‘JC’), Lavandula angustifolia Mill. (‘LA’), Myristica fragrans Houtt. (‘MF’), Malaleuca viridiflora Sol. ex Gaertn. (‘MV’), Origanum vulgare L. (‘OV’), Pogostemon cablin Benth. (‘PC’), Pimenta racemosa (Mill.) J. W. Moore. (‘PR’) and anethol were purchased from EUMA (Bs. As., Argentina).
1H NMR spectra were recorded on a Bruker avance II at 300 MHz in CDCl3, in the presence of TMS (0.00 ppm) as the internal standard. 13C NMR spectra were recorded on the same apparatus at 75 MHz with CDCl3 as solvent and reference (76.9 ppm); 13C NMR assignments were made on the basis of chemical shifts and proton multiplicities (COSY 1H–1H, HSQC, and HMBC).
GC-MS was performed using an Agilent model 7890B Gas Chromatograph coupled to Agilent model 5977A Mass Spectrometer. Column: HP-5MS UI, 30 m × 0.25 mm, 0.25 μm film thick.
Mass spectra were recorded on a Bruker micrOTOF-Q II spectrometer (Bruker-Daltonics). MS parameters: source type: ESI, ion polarity: positive, set nebulizer: 0.4 bar, set dry heater: 180 °C, set dry gas: 4.0 L min−1, set capillary: 4500 V, set end plate offset: −500 V, set collision cell RF: 150.0Vpp.
Searching strategy for functional groups in the dictionary of natural products
To narrow the search only to components of essential oils contained in the DNP, words like ‘oil’, ‘essence’ and ‘essential’ were written in the field ‘source/synthesis’ in addition to drawing the desired structures. The total number of molecules that are essential oil components contained in the DNP is equal to 1507.The number of structures containing non-aromatic double bonds was calculated combining searches as follows: ‘α NOT β = γ; γ OR δ = ε’. Where α represents the number of molecules in the DNP that contain at least one double bond (aromatic or non-aromatic) in their structure, β represents the number of molecules in the DNP that contain at least one aromatic ring in their structure, and δ represents the number of molecules in the DNP that contain at least one aromatic double bond and one non-aromatic double bond in their structure. β was subtracted from α to obtain γ that represents the number of molecules in the DNP that contain only non-aromatic double bonds in their structure. Finally, γ was combined with δ to give ε, which represents the total number of molecules in the DNP that contain at least one non-aromatic double bond in their structure. α = 1178 structures, β = 261 structures, γ = 917 structures, δ = 119 structures, ε = 1036 structures. The average frequency of molecules containing each functional group was standardized to 1507. Bromine moiety was searched within the total number of DNP structures by drawing of ‘Br–C’ in the plot window.
Experimental procedure for the preparation of the BEOs
To a solution of the EO (1 g) in ethyl acetate (50 mL) at −78 °C, bromine (188 μL, 1.1 mmol) was added dropwise, and the reaction was stirred for 2 h at −5 °C. A solution of 10% Na2S2O3 (20 mL) was added, and the reaction mixture was stirred for 30 min. The organic material was then extracted with ethyl acetate (3 × 35 mL), and the combined organic phases were dried over Na2SO4 and evaporated under reduced pressure. Yields: MV = 121%, OV = 142%, FV = 154%, PC = 154%, CC = 159%, JC = 164%, CA = 166%, CL = 169%, COS = 171%, MF = 174%, AAL = 179%, CO = 183%, PR = 185%, CV = 188%, CYC = 197%, LA = 197%, CM = 245%.Chemical impact evaluation
CG-MS were transformed to a two-dimensional format, and statistically processed (PCA) using the Bruker Daltonics ProfileAnalysis 2.0 software. Rectangular bucketing was performed using the following bucketing parameters: the total retention time (RT) range from 5 to 60 minutes, the total mass range from to 500 m/z, delta [minutes] = 0.1 and delta (m/z) = 0.1. PCA of the data were conducted using a MATLAB written code, TOMCAT.40 The routines were developed under MATLAB 7.0 (Release 13).
The comparison of the chromatograms and the m/z and retention time histogram was performed using MZmine software through the RANSAC aligner algorithm prior preprocessing.41 The concentration used for GC-MS of the essential oils was 5 mg mL−1.
Biological impact evaluation
:ethyl acetate (90:10). Once the solvent was completely removed, the plates were subjected to XO autographic assay. A modified protocol reported by Ramallo et al.35 was applied. In this, the agar concentration was modified in order to achieve a thicker agar layer to allow its subsequent manipulation during BIOMSID. Thus, 1% w/v agar final concentration was employed. Each XO biological profile was compared with a riboflavin control35 to check that the observed halos were a true XO inhibition and not an antioxidant activity.BIOMSID analysis
:EtOAc (80:20) and toluene:EtOAc (93:7) respectively. Each TLC was examined for XO activity and gel samples from inhibition zones and background zones were extracted as was suggested in Ramallo et al.36 Each gel portion was extracted with dichloromethane (3 × 1 mL), the solutions were dried and dissolved in methanol (3 mL). The resulting solutions were infused directly into the ESI chamber at a rate of 180 μL min−1 during 1 minute for HRMS analysis. Previous baseline correction (flatness = 0.95), the spectra were exported in -ASCII format, with Data Analysis Version 4.0 SP1. The mass lists, conformed by m/z and respective intensity values, were generated with Peak finder Apex algorithm (Data Analysis Version 4.0 SP1) with the following parameters: peak width (FWHM) = 0.04, S/N threshold = 6.5, relative intensity threshold (base peak) = 0, absolute intensity threshold = 0.Isolation of compound 1
The modified F. vulgare essential oil (330 mg) was chromatographed on silica gel gradient from hexane (100%) to hexane:EtOAc (90:10).
The fractionation was guided through XO autography. (RS)-2-Bromo-1-(4-methoxyphenyl) propan-1-one (C10H11BrO2) (1): yellow oil, 1H NMR (300 MHz; CDCl3) δ = 1.88 (d, J = 7 Hz, 3H, CH3), 3.87 (s, 3H, ArOCH3), 5.25 (q, J = 7 Hz, 1H, CHBr), 6.95 (d, J = 8 Hz, 2H, ArH), 8.00 (d, J = 8 Hz, 2H, ArH); 13C NMR (75 MHz; CDCl3) δ = 20.24, 41.44, 55.55, 113.97, 126.90, 131.30, 163.94 and 192.00. ESI-HRMS [M + H]+ exact mass calc. 243.0015, found: 243.0016, error = 0.2 ppm and [M + Na]+ exact mass calc. 264.9835, found: 264.9833, error = 0.6 ppm.
Synthesis of compound 1
To a solution of the anethol (4 mmol) in ethyl acetate (30 mL) at −78 °C, bromine (230 μL, 4.45 mmol) was added dropwise, and the reaction was stirred for 2 h at 0 °C. A solution of 10% w/v Na2S2O3 (60 mL) was added, and the reaction mixture stirred for 30 min. The organic material was then extracted with ethyl acetate (3 × 30 mL), and the organic phases dried over Na2SO4 and evaporated under reduced pressure. Yield 3.9%.Biological activity quantification
IC50 against XO was measured using a 96-well microplate assay based on a method previously reported.43 Aliquots of ½ seriated DMSO dilutions (10 μL) starting in 45 mM for allopurinol, and compounds 1 and 2, were added on each well (3.7% well final concentration DMSO). The controls contained DMSO instead of compound solutions. Percentage of inhibition was calculated by comparing the rates for each sample to the control and the IC50 was estimated by using GraphPad Prism 5.01 (GraphPad Software, CA, USA).Conclusions
Using a series of essential oils as starting material, we demonstrated that the transformation of double bonds (a highly common group in natural skeletons) is a promising strategy to generate diversified mixtures of natural products.Considering the average numbers, the applied reaction protocol transforms more than 90% of the components of the starting natural mixtures, generating approximately two products from each natural precursor molecule. The reaction increases the molecular weight and decreases the polarity of the molecules population. Assuming that the starting pool of EOs is not redundant in composition, in this set of experiments 781 natural molecules were transformed to produce 1756 unnatural molecules.
The unsupervised multivariate analysis separated the samples in natural and brominated, and indicates that the transformation of double bonds is an important factor for such discrimination, without previous knowledge of the detailed chemical composition of any of the samples.
The use of a TLC autography assay allowed the quick evaluation of the effect of the reaction on bioactivity, spotting the mixtures where the effect was highest. Coupling the assay to HRMS linked the observed XO inhibition to an organobromine compound before any purification step. Bioguided fractionation led to the isolation of (RS)-2-bromo-1-(4-methoxyphenyl) propan-1-one (1). This compound was generated from the inactive natural precursor anethole and showed inhibition properties similar to allopurinol.
Besides the isolation of this particular xanthine oxidase inhibitor, the results illustrate how the combination of a few analytical tools can accelerate the generation, the chemical and biological analyses and the fast identification of bioactive natural products derivatives.
Compound 1 was generated as a minor component of one of 34 complex mixtures (it represented 1.28% of the chromatogram total area of the F. vulgare BEO), however the described combination of tools has paved the way to its characterization.
Acknowledgements
Financial support for this work was provided by FONCYT (PICT2011-0918), CONICET (PIP 695) and Universidad Nacional de Rosario to R. L. F. PG Thank CONICET for her fellowship. IAR, MOS and RLEF are CONICET researchers.Notes and references
- J. Clardy and C. Walsh, Nature, 2004, 432, 829–837 CrossRef CAS PubMed.
- E. C. Barnes, R. Kumar and R. A. Davis, Nat. Prod. Rep., 2016, 33, 372–381 RSC.
- S. Rizzo and H. Waldmann, Chem. Rev., 2014, 114, 4621–4639 CrossRef CAS PubMed.
- K. C. Morrison and P. J. Hergenrother, Nat. Prod. Rep., 2014, 31, 6–14 RSC.
- M. E. Maier, Org. Biomol. Chem., 2015, 13, 5302–5343 CAS.
- K. Tomohara, T. Ito, N. Hasegawa, A. Kato and I. Adachi, Tetrahedron Lett., 2016, 57, 924–927 CrossRef CAS.
- T. Wu, C. Jiang, L. Wang, S. L. Morris-Natschke, H. Miao, L. Gu, J. Xu, K.-H. Lee and Q. Gu, J. Nat. Prod., 2015, 78, 1593–1599 CrossRef CAS PubMed.
- S. N. Lopez, I. A. Ramallo, M. G. Sierra, S. A. Zacchino and R. L. Furlan, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 441–444 CrossRef CAS PubMed.
- T. Kawamura, K. Matsubara, H. Otaka, E. Tashiro, K. Shindo, R. C. Yanagita, K. Irie and M. Imoto, Bioorg. Med. Chem., 2011, 19, 4377–4385 CrossRef CAS PubMed.
- H. Kikuchi, K. Sakurai and Y. Oshima, Org. Lett., 2014, 16, 1916–1919 CrossRef CAS PubMed.
- Z. Lin, X. Ma, H. Wei, D. Li, Q. Gu and T. Zhu, RSC Adv., 2015, 5, 35262–35266 RSC.
- I. A. Ramallo, M. O. Salazar, L. Mendez and R. L. E. Furlan, Acc. Chem. Res., 2011, 44, 241–250 CrossRef CAS PubMed.
- L. Méndez, M. O. Salazar, I. A. Ramallo and R. L. E. Furlan, ACS Comb. Sci., 2010, 13, 200–204 CrossRef PubMed.
- M. O. Salazar, I. A. Ramallo, O. Micheloni, M. G. Sierra and R. L. Furlan, Bioorg. Med. Chem. Lett., 2009, 19, 5067–5070 CrossRef CAS PubMed.
- I. A. Ramallo, M. G. Sierra and R. L. Furlan, Med. Chem., 2012, 8, 112–117 CrossRef CAS.
- Z.-R. Zhang, J.-H. Li, S. Li, A.-L. Liu, P.-M. Hoi, H.-Y. Tian, W.-C. Ye, S. M.-Y. Lee and R.-W. Jiang, PLoS One, 2014, 9, e100416 Search PubMed.
- K. L. Kirk, Org. Process Res. Dev., 2008, 12, 305–321 CrossRef CAS.
- S. M. Johnson, S. Connelly, I. A. Wilson and J. W. Kelly, J. Med. Chem., 2008, 51, 6348–6358 CrossRef CAS PubMed.
- G. W. Gribble, in Naturally Occurring Organohalogen Compounds - A Comprehensive Update, Springer Vienna, Vienna, 2010, pp. 9–348 Search PubMed.
- T. Iwagawa, M. Kaneko, H. Okamura, M. Nakatani and R. W. M. van Soest, J. Nat. Prod., 1998, 61, 1310–1312 CrossRef CAS PubMed.
- T. M. Zabriskie, J. A. Klocke, C. M. Ireland, A. H. Marcus, T. F. Molinski, D. J. Faulkner, C. Xu and J. Clardy, J. Am. Chem. Soc., 1986, 108, 3123–3124 CrossRef CAS.
- M. A. Rashid, K. R. Gustafson, L. K. Cartner, N. Shigematsu, L. K. Pannell and M. R. Boyd, J. Nat. Prod., 2001, 64, 117–121 CrossRef CAS.
- F. Cafieri, E. Fattorusso and O. Taglialatela-Scafati, J. Nat. Prod., 1998, 61, 122–125 CrossRef CAS PubMed.
- G. W. Gribble, Am. Sci., 2004, 92, 342 CrossRef.
- A. E. Edris, Phytother. Res., 2007, 21, 308–323 CrossRef CAS PubMed.
- M. E. Maffei, J. Gertsch and G. Appendino, Nat. Prod. Rep., 2011, 28, 1359–1380 RSC.
- P. Pacher, A. Nivorozhkin and C. Szabó, Pharmacol. Rev., 2006, 58, 87–114 CrossRef CAS PubMed.
- K. Nepali, G. Singh, A. Turan, A. Agarwal, S. Sapra, R. Kumar, U. C. Banerjee, P. K. Verma, N. K. Satti, M. K. Gupta, O. P. Suri and K. L. Dhar, Bioorg. Med. Chem., 2011, 19, 1950–1958 CrossRef CAS PubMed.
- K. Nepali, A. Agarwal, S. Sapra, V. Mittal, R. Kumar, U. C. Banerjee, M. K. Gupta, N. K. Satti, O. P. Suri and K. L. Dhar, Bioorg. Med. Chem., 2011, 19, 5569–5576 CrossRef CAS PubMed.
- R. Dhiman, S. Sharma, G. Singh, K. Nepali and P. M. S. Bedi, Arch. Pharm., 2013, 58, 87–114 Search PubMed.
- H. Singh, S. Sharma, R. Ojha, M. K. Gupta, K. Nepali and P. M. S. Bedi, Bioorg. Med. Chem., 2014, 24, 4192–4197 CrossRef CAS PubMed.
- S. Shukla, D. Kumar, R. Ojha, M. K. Gupta, K. Nepali and P. M. S. Bedi, Arch. Pharm., 2014, 347, 486–495 CrossRef CAS PubMed.
- H. S. Virdi, S. Sharma, S. Mehndiratta, P. M. S. Bedi and K. Nepali, J. Enzyme Inhib. Med. Chem., 2015, 5, 730–736 CrossRef PubMed.
- Ł. M. Cieśla, M. Waksmundzka-Hajnos, K. A. Wojtunik and M. Hajnos, Phytochem. Lett., 2015, 11, 445–454 CrossRef.
- I. A. Ramallo, S. A. Zacchino and R. L. E. Furlan, Phytochem. Anal., 2006, 17, 15–19 CrossRef CAS PubMed.
- I. A. Ramallo, M. O. Salazar and R. Furlan, Phytochem. Anal., 2015, 26, 404–412 CrossRef CAS PubMed.
- F. W. McLafferty and F. Tureček, Interpretation of Mass Spectra, University Science Books, Mill Valley, 1993 Search PubMed.
- A.-U. Rahman, M. I. Choudhary and W. J. Thomsen, Bioassay Techniques for Drug Development, Taylor & Francis, Amsterdam, 2001 Search PubMed.
- M. R. Viant, Biochem. Biophys. Res. Commun., 2003, 310, 943–948 CrossRef CAS PubMed.
- M. Daszykowski, S. Serneels, K. Kaczmarek, P. Van Espen, C. Croux and B. Walczak, Chemom. Intell. Lab. Syst., 2007, 85, 269–277 CrossRef CAS.
- T. Pluskal, S. Castillo, A. Villar-Briones and M. Oresic, BMC Bioinf., 2010, 11, 395 CrossRef PubMed.
- M. Feher and J. M. Schmidt, J. Chem. Inf. Comput. Sci., 2002, 43, 218–227 CrossRef PubMed.
- Y.-H. Chu, C.-J. Chen, S.-H. Wu and J.-F. Hsieh, J. Agric. Food Chem., 2014, 62, 3742–3749 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental results and DNP search strategy. See DOI: 10.1039/c6ra05373d |
| This journal is © The Royal Society of Chemistry 2016 |