DOI:
10.1039/C6RA05320C
(Paper)
RSC Adv., 2016,
6, 44080-44086
Asymmetric dendrimers with improved electro-optic performance: synthesis and characterization†
Received
29th February 2016
, Accepted 18th April 2016
First published on 20th April 2016
Abstract
A new series of electro-optical dendrimers with asymmetric configurations was synthesized through a Cu-(I) catalyzed Huisgen reaction. The asymmetric dendrimers exhibited weaker chromophore dipole–dipole aggregations along with an improved response to the applied poling field than traditional dendrimers with the same type of chromophores. In order to systematically investigate the influence of the asymmetric structure on the physical properties, UV-vis spectroscopy and DFT calculations were carried out. Due to the asymmetric configurations, the obtained dendrimers exhibited weak dipole–dipole interactions compared with dendrimers with a flexible linker, furthermore they obtained a better alignment in the poling process. The asymmetric dendrimer AD-2 exhibited a 36 pm V−1 EO coefficient measurement, which was higher than the other two dendrimers and guest–host polymeric materials using the same chromophores.
1. Introduction
Organic electro-optical (EO) materials have been proven to be promising candidates for the development of photonic integrated devices over the past two decades.1–6 Lots of materials are synthesized to improve the macro EO activity. For most organic EO materials, increasing the doping level of chromophores within a rational range will enhance the EO performance. When the concentration of chromophores is over the optimum level, it can lead to strong dipole–dipole interactions and optical loss due to possible phase separation.7,8 Thus, how to efficiently avoid deleterious aggregation effects and translate the microscopic nonlinear optical activity (β) into macroscopic EO activities have already become the major focus for improving modern EO materials.9–13
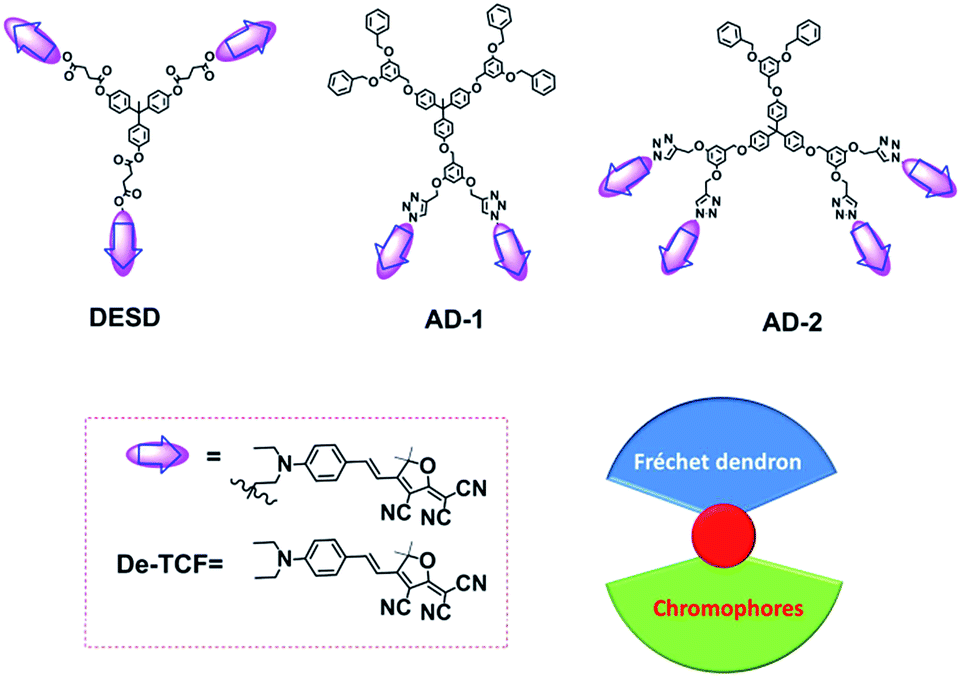
In recent years, multichromophore dendritic EO materials have been proven as an effective approach to inhibit interchromophore electrostatic interactions as well as to enhance the poling efficiency attributed to spherical configurations.14,15 Based on the classic dendrimer’s structural motif (Scheme 1), the component chromophores are covalently attached to the dendrimer branches and can rotate freely if the linker is sufficiently long. However, if the chromophore density is too high this can lead to increased material conductivity, thus limiting the effectiveness of the poling field in reorienting the chromophores.2 Symmetric multichromophore dendrimers (MCDs) may imply that the component chromophore dipole moments effectively cancel resulting in a low net dipole moment. Additionally, reorientation under a poling field is inhibited in symmetric MCDs (DESD; Scheme 1) which have low net molecular dipole moments. In this article, we propose two kinds of novel EO multichromophore dendrimers with asymmetric configurations that were synthesized via a Huisgen reaction according to previous reports.16–19 These materials were developed and tested in an effort to improve both poling efficiency and control of material conductivity in organic EO materials.
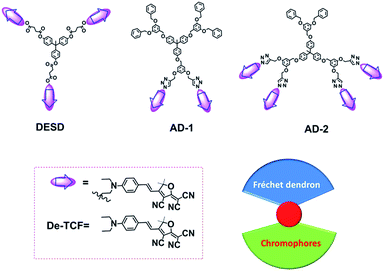 |
| | Scheme 1 The structures of dendrimers and chromophores. | |
2. Results and discussion
2.1 Synthesis
Scheme 1 shows the asymmetric dendrimer structures, AD-1 and AD-2. Included in the dendrimer backbone are 3,5-dioxybenzyl ether units which possess enhanced rigidity and stability. In order to improve the molecular response to the applied poling field and control the material conductivity, the chromophores were attached to a portion of the branches (1/3 or 2/3) using the Huisgen reaction, leaving the remaining branches functionalized with Fréchet dendrons as isolation groups.20 During this process, the dendrimers maintained ellipsoidal configuration by controlling the generation of different branches.
As shown in Scheme 2, the synthesis of AD-1 and AD-2 resulted in high yields (final step: >80%). Electron donor 2 was obtained by 4-((2-hydroxyethyl)(methyl)amino)-benzaldehyde (1) reacting with diphenylphosphoryl azide (DPPA), following a safe, high-efficiency route to turn the hydroxyl group on (1) into an azide group. By controlling the amount of 1,1,1-tris(4-hydroxy-phenyl)ethane, compounds 4 and 6 can be acquired using different ratios (2 eq. and 0.5 eq., respectively). A combination of copper sulfate + sodium ascorbate was adopted as a catalyzer, instead of copper-(I) + amine which acts as a strong nucleophile causing acceptor ring opening, thereby causing the decay of chromophore.
 |
| | Scheme 2 Synthesis routes for two asymmetric dendrimers. | |
In addition, guest–host polymer film P-1 was prepared by blending the chromophore De-TCF into PMMA (Mw ∼ 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000), using the concentrations as for AD-1. After stirring for one day, there was still a portion of chromophores remaining solid which was removed via filtration. In order to investigate the chromophore concentration in P-1, we prepared a series of polymer solutions with gradient concentrations (wt: 5%, 10%, 15% and 20%) to find the saturated point. A small quantity of unsolvable chromophores were found in the 20% wt polymer solution, which implied the real concentration of P-1 should be below 20% wt.
000), using the concentrations as for AD-1. After stirring for one day, there was still a portion of chromophores remaining solid which was removed via filtration. In order to investigate the chromophore concentration in P-1, we prepared a series of polymer solutions with gradient concentrations (wt: 5%, 10%, 15% and 20%) to find the saturated point. A small quantity of unsolvable chromophores were found in the 20% wt polymer solution, which implied the real concentration of P-1 should be below 20% wt.
2.2 1H-NMR and infrared spectra
1H-NMR and FTIR spectra were used to identify the chemical structure of the obtained dendrimers. Fig. 1 shows the 1H-NMR spectra for AD-1 and AD-2 together with their precursors. The signals at 1.69 ppm for AD-1 and AD-2 are attributed to the protons in the methyl groups of the tricyanofuran (TCF) acceptors which feature negligible displacement with those of chromophore N3-TCF. In the spectra of dendrimers 5 and 8, alkynyl protons showed triplet peaks at 2.49 ppm. After the Huisgen reaction, the peaks of the alkynyl protons disappeared completely, which indicates that the starting materials efficiently converted into desired dendrimers AD-1 and AD-2. At the same time, a new broad single peak appeared at 5.09 ppm which belonged to 1,2,3-triazole.20 Owing to the transformation of azide groups in N3-TCF, the signals of two methylene protons in the donor moved from 3.57 and 3.69 triplet peaks to 3.94 and 4.57 singlet peaks in AD-1 and AD-2. Except for the signals of vinyl protons and aromatic protons, which were overlapped by the proton signals of the dendrimer’s backbone, the other signals of N3-TCF could be easily observed in the 1H-NMR spectra. All chemical shifts were consistent with the proposed dendrimers’ structure. The measured ratios were in keeping with the ratios of the proposed structures, which proved the content of chromophores in the dendrimers can be controlled accurately, and the reproducibility of the dendrimers’ property is guaranteed. The FT-IR spectra (Fig. 2) show a strong absorption peak near 2222 cm−1 indicating the presence of a nitrile group, and typical bands of aromatic rings at 1597 cm−1 and 1523 cm−1.
 |
| | Fig. 1 1H-NMR spectra comparison for the dendrimers. | |
 |
| | Fig. 2 Infrared spectra for AD-1 and AD-2. | |
2.3 DSC curves and UV-vis spectra analyses
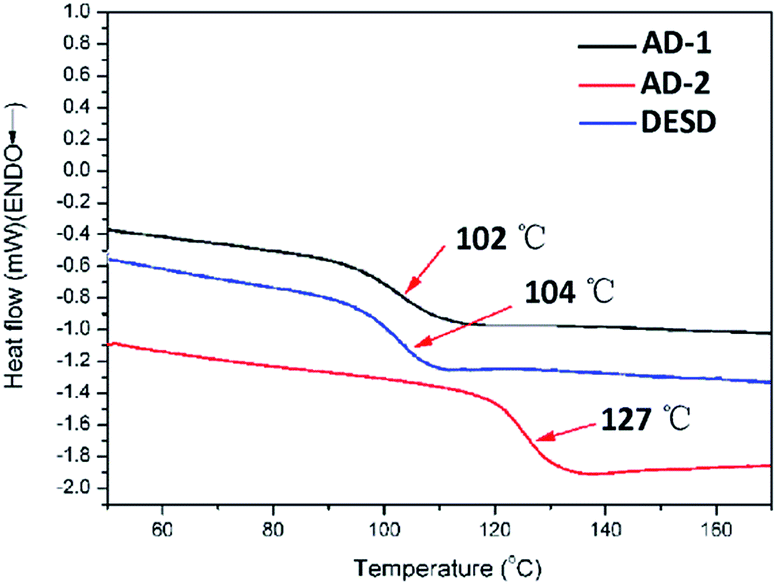
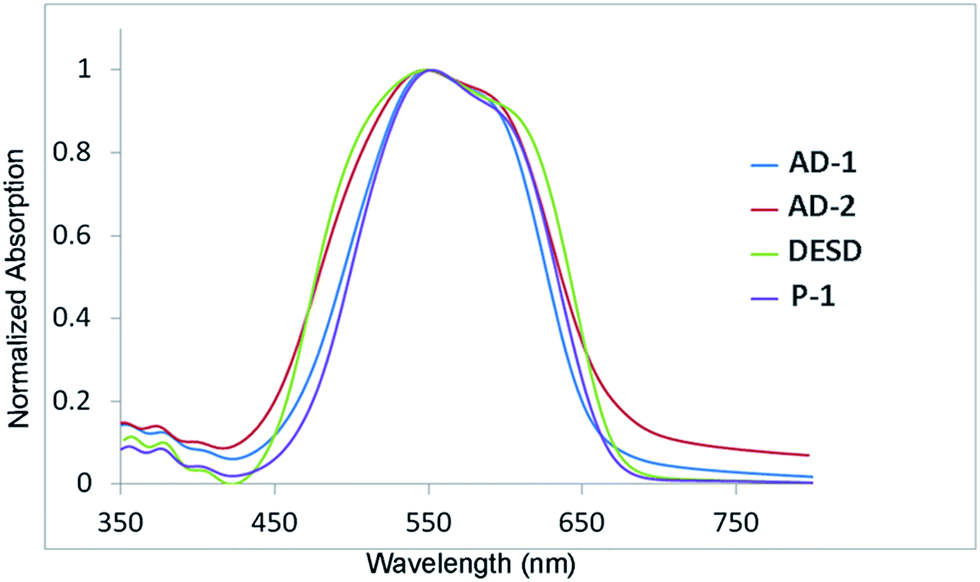
AD-1 and AD-2 exhibited good solubility in 1,1,2-trichloroethylene and an excellent film-forming property. High-quality, smooth, homogeneous, and highly transparent films were obtained via spin-coating. To systematically examine the improvement of the materials properties brought by the asymmetric dendritic structure, a traditional dendrimer DESD (Scheme 1) was synthesized and characterized in accordance with the methods used for AD-1 and AD-2. In the thermoanalysis, both AD-1 and AD-2 began to decompose at 250 °C which is the same as the chromophore De-TCF, indicating that the decomposition of AD-1 and AD-2 at this temperature was induced by the decomposition of the chromophores.21 AD-2 exhibited the highest Tg (127 °C) value compared with AD-1 and DESD which had Tg values of 102 °C and 104 °C, as shown in Fig. 3. UV-vis spectra were recorded to locate the charge transfer (CT) peak of the chromophore as well as to examine possible aggregation behaviour, typically indicated by an additional low-energy (high-wavelength) absorption near the CT peak.6,12,22–24 We recorded the UV-vis spectra in chloroform (Fig. 4) and films (Fig. 5). The synthesised dendrimers and chromophore exhibited a similar pi/pi* intramolecular charge-transfer (ICT) absorption band in the visible region (dendrimers: 10−6 mol L−1; De-TCF: 10−5 mol L−1). With the increase of concentration, there are no apparent changes for the shape and location of the absorption peaks in chloroform at the concentration range in which the spectrometer can measure the UV-vis absorption. By testing the different concentrations of these three chromophores in CHCl3, we observe no significant dipole–dipole interactions at low concentration.
 |
| | Fig. 3 The DSC curves for the dendrimers. | |
 |
| | Fig. 4 UV-vis spectra in chloroform with different concentrations. | |
 |
| | Fig. 5 UV-vis spectra in films. | |
Fig. 4 shows the absorption curves of four films. Generally, the characteristic absorption band of the film will be broader than in the solutions because of material inhomogeneity and chromophore interactions in the film.25–27 The nature of chromophore aggregation that results in the noticeable bathochromic absorption shoulder appearing in the thin film spectra have been previously attributed to the occurrence of inhomogeneous broadening and solvatochromism of the electronic absorption characteristics associated with dye–dye and dye–polymer interactions. The intrinsic nature of the bathochromic absorption shoulders appearing in the films of all four materials generally are interpreted as the nanoscale acentric J-aggregation between chromophores and centrosymmetric aggregation.26,28–32 To a certain extent, the absorption strength of the shoulder and the peak width of the CT-band reflect the degree of chromophore dipole–dipole interaction for materials containing the same type of chromophores.28,33,34 For example, DESD showed the widest CT-band and the most exaggerated shoulder. On the contrary, AD-2 exhibited a narrow CT-band and a relatively weak shoulder. These results indicate that aggregation effects are more prominent in DESD than in AD-2 or AD-1. Furthermore, these results indicate that the flexible linkers performed poorly in preventing chromophore aggregations via site-isolation of each active unit within the internal free volume created by the dendrimer. Although P-1 contained a lower chromophore concentration (≤20%), it also displayed similar shoulders in the right of the CT-band compared with AD-1 (Table 1).
Table 1 UV-vis data and r33 values
| |
λmaxa (nm) |
λmaxb (nm) |
r33 (pm V−1) |
| Measured in films. Measured in chloroform. |
| AD-1 |
553 |
539 |
23 |
| AD-2 |
550 |
536 |
36 |
| DESD |
545 |
563 |
19 |
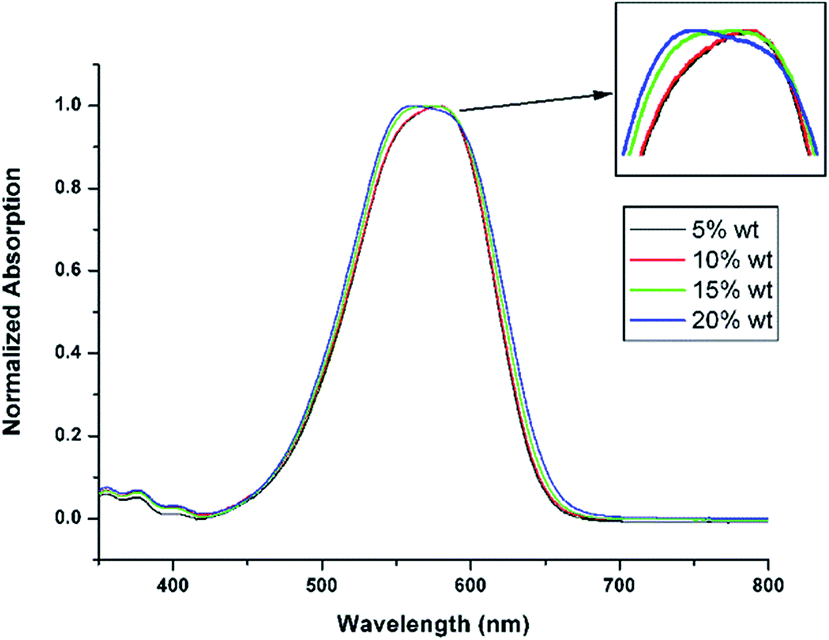
In order to investigate the dye dispersion behaviour in the PMMA matrix, we prepared a series of guest–host polymer films using the chromophore De-TCF and PMMA, and measured their UV-vis spectra (Fig. 6). With the increasing chromophore concentration, there is an apparent blue-shift for the maximum absorption peaks (λmax: 578 nm/5% wt; 576 nm/10% wt; 572 nm/15% wt; 560 nm/20% wt), which should be attributed to the H-aggregations of the chromophores. At the same time, the CT-bands become slightly broader. According to the exciton theory, the H-aggregates will be characterized by hypsochromic (blue-shifted) and J-aggregates by a bathochromic (red-shifted) absorption with respect to the monomer UV-vis absorption band. Spectral broadening has been widely observed in NLO chromophore-doped guest–host materials, and has been attributed to the inhomogeneous broadening and solvatochromism of the electronic absorption characteristics associated with dye–dye and dye–polymer interactions.35
 |
| | Fig. 6 UV-vis spectra for De-TCF in PMMA. | |
2.4 DFT calculations and EO performances
To obtain further understanding of the effect of the asymmetric structure on molecular movement and rotation, density functional theory (DFT) calculations using Gaussian 09 were carried out on three dendrimers at the PM3 level, employing the split valence 3-21g basis set.36 All calculations converged to a RMS error in the density matrix of <10−11 au. Geometry optimized structures (Fig. 7) at this level of theory for AD-1 and AD-2 possess well dendronized configurations which are close to ellipsoidal and are proposed to be effective in inhibiting anti-parallel dipole–dipole aggregation. On the contrary, DESD showed a rod-like optimized structure and two chromophores aggregating together. Rod-like structures have poor performance in isolating chromophores and, thus, are undesirable for this study.
 |
| | Fig. 7 Optimized configurations of the dendrimers. | |
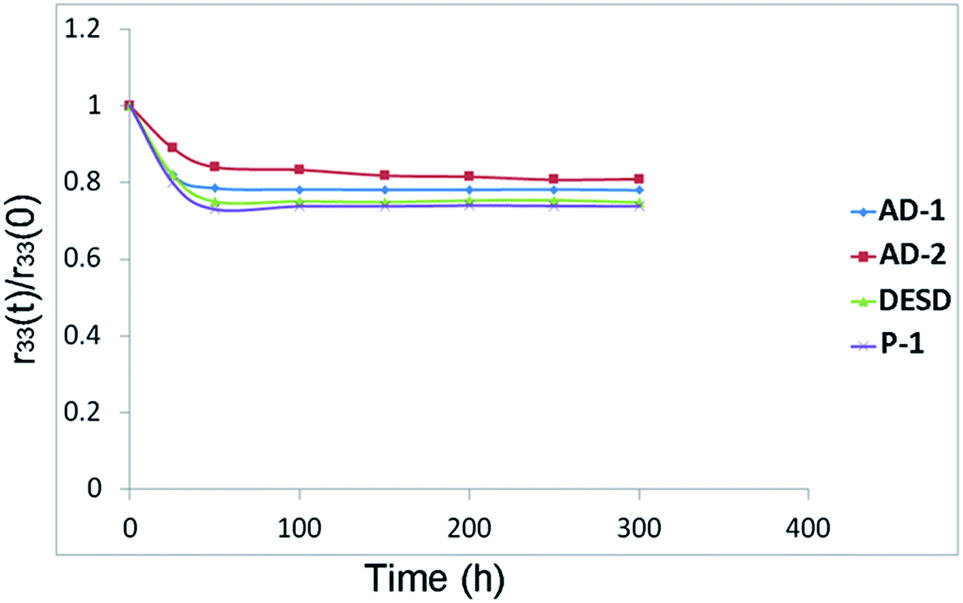
The EO coefficients of AD-1 and AD-2 were 23 pm V−1 and 36 pm V−1 (1310 nm), respectively. Following the same process of preparation, the maximum r33 exhibited by poled DESD-films was 19 pm V−1 for a doping concentration of nearly 63%. Despite this being the maximum achieved r33 for this material, the high chromophore doping concentration likely induced severe dipole–dipole interactions, thus partly accounting for reduction in EO performance of DESD-films. The EO activity obtained by the poled-film AD-2 was more than five times larger than the chromophore-doping film P-1, for which the maximum r33 values were approximately 7 pm V−1. The great improvement of EO activities for AD-2 indicates that the intermolecular electrostatic interactions were effectively suppressed in this polymer system and these NLO chromophores could be well isolated on the polymer chain. After 300 h at 75 °C, the EO activities can be retained above 81% of the initial data for AD-2, which were higher than DESD (75%) and P-1 (72%), as shown in Fig. 8. The improvement of thermal stability for EO activity should be attributed to the rigid structure which helped in restraining chromophore depolarization.
 |
| | Fig. 8 Temporal stability for poled films. | |
3. Experimental details
3.1 General procedures
All chemicals are commercially available and were used without further purification unless otherwise stated. N,N-Dimethyl formamide (DMF) was distilled over calcium hydride and stored over molecular sieves (pore size 3 Å). Acetone was dried with anhydrous MgSO4, then distilled and stored over molecular sieves (pore size 3 Å). The 2-dicyanomethylene-3-cyano-4-methyl-2,5-dihydrofuran (TCF) acceptor was prepared according to the literature.37 Compound 2 and N3-TCF were prepared according to the literature.17 Compounds 3 and 5 were prepared according to the literature.20 DESD was synthesized according to a previous report.19 The PMMA was bought from Ouhe Corporation, Beijing. The Mw is about 10000. The Tg is 105 °C. TLC analyses were carried out on 0.25 mm thick precoated silica plates and spots were visualized under UV light. Chromatography on silica gel was carried out on Kieselgel (200–300 mesh). 1H and 13C NMR spectra were obtained using an Advance Bruker 400M (400 MHZ) NMR spectrometer (tetramethylsilane as internal reference). The MS spectra were obtained using MALDI-TOF (Matrix Assisted Laser Desorption/Ionization of Flight) on a BIFLEXIII (Broker Inc.) spectrometer. The UV-vis experiments were performed on a Cary 5000 photospectrometer. The TGA was determined using a TA5000-2950TGA (TA co) with a heating rate of 10 °C min−1 under the protection of nitrogen. Atomic Force Microscopy (AFM) was carried out using a multimode 8 Bruker. DFT calculations using Gaussian 09 were carried out on three dendrimers at the PM3 level, employing the split valence 3-21 basis set. All calculations converged to a RMS error in the density matrix of <10−11 au.
3.2 Synthesis of compound 4
To a stirred solution of compound 3 (3.38 g, 10 mmol) and methyl 1,1,1-tris(4-hydroxyphenyl)ethane (3.06 g, 10 mmol) in acetone (300 mL), cesium carbonate (3.26 g, 10 mmol) and 18-crown-6 (0.1 g, 0.4 mmol) were added. The reaction mixture was heated at reflux under nitrogen for 24 h, filtered, evaporated to dryness, and partitioned between water and dichloromethane. The aqueous layer was then extracted with dichloromethane (2 × 100 mL), and the combined extracts were dried and evaporated to dryness. The crude material was then purified by column chromatography using dichloromethane and methanol as the eluent to give compound 4 as a colorless solid (5.29 g, 87.1%). 1H NMR (400 MHz, DMSO) δ: 9.23 (s, 2H, –OH), 7.42 (d, 4H, Ar-H), 7.37 (t, 4H, Ar-H), 7.31 (d, 2H, Ar-H), 6.92 (d, 2H, Ar-H), 6.86 (d, 2H, Ar-H), 6.81 (d, 4H, Ar-H), 6.69 (s, 2H, Ar-H) 6.66 (d, 4H, Ar-H), 6.62 (s, 1H, Ar-H), 5.06 (s, 4H, –CH2), 4.96 (s, 2H, –CH2), 1.97 (s, 3H, –CH3). 13C NMR (101 MHz, CDCl3) δ: 156.15, 151.78, 144.25, 141.58, 140.54, 137.57, 129.36, 125.65, 114.67, 112.83, 107.10, 102.00, 78.46, 75.69, 69.89, 56.12, 50.78, 30.83. MALDI-TOF (M+): calcd: 608.72; found: 608.69.
3.3 Synthesis of dendrimer 5
The procedure for compound 4 was followed to prepare dendrimer 5 as a colorless solid (91.3%). 1H NMR (400 MHz, CDCl3) δ: 7.36 (m, 10H, Ar-H), 6.98 (dd, J = 8.9, 2.3 Hz, 6H, Ar-H), 6.86–6.81 (m, 6H, Ar-H), 6.68 (t, J = 2.6 Hz, 6H, Ar-H), 6.56 (d, J = 2.1 Hz, 3H, Ar-H), 5.03 (s, 4H, –CH2), 4.97 (d, J = 6.7 Hz, 6H, –CH2), 4.67 (d, J = 2.4 Hz, 8H, –CH2), 2.50 (t, J = 2.4 Hz, 4H, alkyne-H), 2.10 (s, 3H, –CH3). 13C NMR (101 MHz, CDCl3) δ: 160.29, 158.99, 156.80, 142.26, 139.94, 136.93, 129.77, 128.71, 128.13, 127.67, 114.18, 107.00, 106.54, 101.85, 78.45, 75.85, 56.08, 30.95. MALDI-TOF (M+): calcd: 1005.16; found: 1005.14.
3.4 Synthesis of compound 6
To a stirred solution of compound 3 (3.38 g, 10 mmol) and methyl 1,1,1-tris(4-hydroxyphenyl)ethane (1.52 g, 5 mmol) in acetone (300 mL), cesium carbonate (3.26 g, 10 mmol) and 18-crown-6 (0.1 g, 0.4 mmol) were added. The reaction mixture was heated at reflux under nitrogen for 24 h, filtered, evaporated to dryness, and partitioned between water and dichloromethane. The aqueous layer was then extracted with dichloromethane (2 × 100 mL), and the combined extracts were dried and evaporated to dryness. The crude material was then purified by column chromatography using dichloromethane and methanol as the eluent to give compound 6 as a colorless solid (4.03 g, 88.6%). 1H NMR (400 MHz, CDCl3) δ 7.44–7.34 (m, 16H, Ar-H), 7.31 (t, J = 7.0 Hz, 4H, Ar-H), 6.98 (d, J = 8.8 Hz, 4H, Ar-H), 6.94 (d, J = 8.6 Hz, 2H, Ar-H), 6.82 (d, J = 8.8 Hz, 4H, Ar-H), 6.70 (d, J = 8.6 Hz, 2H, Ar-H), 6.68 (s, 4H, Ar-H), 6.56 (s, 2H, Ar-H), 5.03 (s, 8H, CH2), 4.96 (s, 4H, CH2–H), 2.10 (s, 3H, CH3). 13C NMR (101 MHz, CDCl3, ppm): δ = 157.03, 152.83, 146.41, 142.38, 141.87, 139.97, 129.86, 129.71, 114.69, 114.23, 107.10, 102.00, 78.46, 75.69, 69.89, 56.12, 50.78, 30.83. MALDI-TOF (M+): calcd: 910.39; found: 910.37.
3.5 Synthesis of dendrimer 8
The procedure for compound 4 was followed to prepare dendrimer 8 as a colorless solid (91.3%). 1H NMR (400 MHz, CDCl3): δ = 7.45–7.31 (m, 20H, Ar-H), 7.02 (dd, J = 8.9, 2.2 Hz, 6H, Ar-H), 6.86 (d, J = 8.9 Hz, 6H, Ar-H), 6.71 (d, J = 2.1, 6H, Ar-H), 6.59 (t, J = 2.1 Hz, 3H, Ar-H), 5.05 (s, 8H, CH2), 4.99 (d, J = 6.0 Hz, 6H, CH2), 4.68 (d, J = 2.4 Hz, 4H, CH2), 2.52 (t, 2H, alkyne-H), 2.08 (s, 3H, CH3). 13C NMR (101 MHz, CDCl3) δ = 160.30, 159.00, 156.93, 142.24, 139.78 (s), 136.95, 129.78, 128.73, 128.14, 127.69, 114.18, 107.01, 106.56, 75.86, 70.26, 56.09. MALDI-TOF (M+): calcd: 1108.46; found: 1108.38.
3.6 Synthesis of dendrimer AD-1
A solution of the asymmetric dendrimer 8 (0.11 g, 0.1 mmol), chromophore N3-TCF (0.093 g, 0.24 mmol), sodium ascorbate (2 mg, 12 μmol) and CuSO4 (1 mg, 6 μmol) in acetone (25 mL) was stirred at room temperature for 48 h. After evaporation of the solvents, the crude product was purified by column chromatography, eluted with a 9:1 mixture of dichloromethane and methanol, to give AD-1 as a dark red solid (94.0% yield). 1H NMR (400 MHz, CDCl3) δ 7.59 (d, 15.9 Hz, 2H, CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH), 7.48 (d, J = 6.9 Hz, 2H, CHCN), 7.44–7.28 (m, 20H, Ar-H), 7.03–6.94 (d, 6H, Ar-H), 6.80 (d, 6H, Ar-H), 6.72 (d, J = 15.9 Hz, 2H, CHCH), 6.67 (d, J = 2.0 Hz, 6H, Ar-H), 6.60–6.53 (m, 6H, Ar-H), 5.11 (s, 4H, CH2), 5.03 (s, 8H, CH2), 4.94 (d, J = 13.5 Hz, 6H, CH2), 4.62 (s, 4H, CH2), 3.98 (s, 4H, CH2), 2.88 (s, 6H, CH3), 2.09 (s, 3H, CH3), 1.72 (s, 12H, CH3). 13C NMR (101 MHz, CDCl3) δ = 177.09, 175.38, 161.01, 160.28, 157.44, 153.05, 149.08, 143.03, 140.72, 137.61, 133.07, 130.49, 129.45, 128.89, 128.40, 123.72, 114.87, 113.10, 110.52, 107.40, 98.09, 70.99, 53.17, 39.59, 27.46. MALDI-TOF (M+): calcd: 1907.82; found: 1907.79. HRMS (ESI) (M+, C119H106N14O11): calcd: 1906.81655; found: 1906.81648.
CH), 7.48 (d, J = 6.9 Hz, 2H, CHCN), 7.44–7.28 (m, 20H, Ar-H), 7.03–6.94 (d, 6H, Ar-H), 6.80 (d, 6H, Ar-H), 6.72 (d, J = 15.9 Hz, 2H, CHCH), 6.67 (d, J = 2.0 Hz, 6H, Ar-H), 6.60–6.53 (m, 6H, Ar-H), 5.11 (s, 4H, CH2), 5.03 (s, 8H, CH2), 4.94 (d, J = 13.5 Hz, 6H, CH2), 4.62 (s, 4H, CH2), 3.98 (s, 4H, CH2), 2.88 (s, 6H, CH3), 2.09 (s, 3H, CH3), 1.72 (s, 12H, CH3). 13C NMR (101 MHz, CDCl3) δ = 177.09, 175.38, 161.01, 160.28, 157.44, 153.05, 149.08, 143.03, 140.72, 137.61, 133.07, 130.49, 129.45, 128.89, 128.40, 123.72, 114.87, 113.10, 110.52, 107.40, 98.09, 70.99, 53.17, 39.59, 27.46. MALDI-TOF (M+): calcd: 1907.82; found: 1907.79. HRMS (ESI) (M+, C119H106N14O11): calcd: 1906.81655; found: 1906.81648.
3.7 Synthesis of dendrimer AD-2
A solution of the asymmetric dendrimer 5 (0.05 g, 0.05 mmol), chromophore N3-TCF (0.093 g, 0.24 mmol), sodium ascorbate (2 mg, 12 μmol) and CuSO4 (1 mg, 6 μmol) in acetone (25 mL) was stirred at room temperature for 48 h. After evaporation of the solvents, the crude product was purified by column chromatography, eluted with a 9:1 mixture of dichloromethane and methanol, to give AD-2 as a dark red solid (91.0% yield). 1H NMR (400 MHz, CDCl3) δ: 7.61 (d, 8H, CHCH + CHCN), 7.48 (d, 8H, Ar-H), 7.43–7.28 (m, 10H, Ar-H), 6.97 (t, 6H, Ar-H), 6.83 (t, 6H, Ar-H), 6.73 (d, 4H, CHCH), 6.68 (d, 4H, Ar–H), 6.63 (s, 2H, Ar–H), 6.58 (d, 9H, Ar–H), 6.46 (s, 2H, Ar–H), 5.30 (s, 4H, –CH2) 5.11 (s, 8H, –CH2), 5.02 (s, 4H, –CH2), 4.93 (d, 6H, –CH2), 4.60 (s, 8H, –CH2), 3.97 (s, 8H, –CH2), 2.87 (s, 12H, –CH3), 2.07 (s, 3H, –CH3), 1.71 (s, 24H, –CH3). 13C NMR (101 MHz, CDCl3) δ: 177.09, 175.38, 161.01, 160.28, 153.05, 149.08, 137.61, 133.07, 130.49, 129.45 (s), 128.89, 128.40, 123.72, 114.87, 113.10, 110.52, 107.40, 98.09, 95.80, 70.99, 62.71, 39.59, 27.46. MALDI-TOF (M+): calcd: 2546.84; found: 2546.81. HRMS (ESI) (M+, C151H132N28O13): calcd: 2545.05287; found: 2545.05285.
3.8 General procedure for preparing films
In order to evaluate their EO activities, the dendritic and polymeric films using 1,1,2-trichloroethane (TCE) as the solvent were prepared to investigate the translating of the microscopic hyperpolarizability into macroscopic EO response (r33). The solution was filtered using a 0.2 μm syringe filter to remove large particulates. Then the solution was spin-coated on indium-tin oxide (ITO) glass substrates. The films were dried in vacuo for 12 h to remove the residual solvent. The thickness of the films was measured with an Ambios Technology XP-1 profilometer. The thickness of the films was about 2.1–2.6 μm, which should be thicker than the films poled in contact poling, to prevent possible film damage (in contact poling, film thickness will be about 1.3–2.0 μm).
4. Conclusion
In conclusion, we have developed a facile route to synthesize a new kind of EO dendrimer with a unique asymmetric structure. Using the Cu-(I) catalyzed Huisgen reaction, the chromophores (N3-TCF) were attached to the dendrimer backbone with high efficiency (final step yield ≥90%). After corona poling, a large EO coefficient of 36 pm V−1 was achieved for AD-2, which was more than twice that of DESD containing the same chromophores. The asymmetric dendrimer (AD-2) also exhibited excellent thermal stability such that these materials retained above 81% after 300 h at 75 °C. The asymmetric EO dendrimers could effectively improve the response of the chromophore to an applied poling field and could be used in EO device fabrication.
Acknowledgements
We are grateful to the National Natural Science Foundation of China (No. 11104284, No. 61101054 and No. U1533121) for the financial support. We also thank Dr Kerry Garrett for many useful comments and manuscript revision.
References
- S. R. Marder and J. W. Ferry, Science, 1994, 263, 1706–1707 CAS
 .
. - L. R. Dalton, P. A. Sullivan and D. H. Bale, Chem. Rev., 2010, 110, 25–55 CrossRef CAS PubMed .
- J. Wu, J. Liu, T. Zhou, S. Bo, L. Qiu, Z. Zhen and X. Liu, RSC Adv., 2012, 2, 1416–1423 RSC .
- H. Huang, G. Deng, J. Liu, J. Wu, P. Si, H. Xu, S. Bo, L. Qiu, Z. Zhen and X. Liu, Dyes Pigm., 2013, 99, 753–758 CrossRef CAS .
- J. Wu, RSC Adv., 2014, 4, 49737–49744 RSC .
- J. Wu, C. Peng, H. Xiao, S. Bo, L. Qiu, Z. Zhen and X. Liu, Dyes Pigm., 2014, 104, 15–23 CrossRef CAS .
- H. Xu, M. Zhang, A. Zhang, G. Deng, P. Si, H. Huang, C. Peng, M. Fu, J. Liu, L. Qiu, Z. Zhen, S. Bo and X. Liu, Dyes Pigm., 2014, 102, 142–149 CrossRef CAS .
- Y. Q. Shi, C. Zhang, H. Zhang, J. H. Bechtel, L. R. Dalton, B. H. Robinson and W. H. Steier, Science, 2000, 288, 119–122 CrossRef CAS PubMed .
- P. A. Sullivan, H. L. Rommel, T. Yoshinari, S. R. Hammond, D. H. Bale, B. C. Olbricht, L. Yi, R. John, B. E. Eichinger and K.-Y. Alex, J. Phys. Chem. B, 2009, 113, 15581–15588 CrossRef CAS PubMed .
- S. J. Benight, L. E. Johnson, B. Robin, B. C. Olbricht, D. H. Bale, P. J. Reid, B. E. Eichinger, L. R. Dalton, P. A. Sullivan and B. H. Robinson, J. Phys. Chem. B, 2010, 114, 11949–11956 CrossRef CAS PubMed .
- P. A. Sullivan and L. R. Dalton, Acc. Chem. Res., 2010, 43, 428–431 CrossRef PubMed .
- J. Wu, S. Bo, W. Wang, G. Deng, Z. Zhen, X. Liu and K. S. Chiang, RSC Adv., 2015, 5, 102108–102114 RSC .
- J. Wu, W. Wang, L. Wang, J. Liu, K. Chen and S. Bo, Mater. Lett., 2015, 164, 636–639 CrossRef .
- P. A. Sullivan, A. J. P. Akelaitis, S. K. Lee, G. McGrew, S. K. Lee, D. H. Choi and L. R. Dalton, Chem. Mater., 2006, 18, 344–351 CrossRef CAS .
- Z. Li, Q. Q. Li and J. G. Qin, Polym. Chem., 2011, 2, 2723–2740 RSC .
- C. R. Becer, R. Hoogenboom and U. S. Schubert, Angew. Chem., Int. Ed. Engl., 2009, 48, 4900–4908 CrossRef CAS PubMed .
- J. Liu, P. Si, X. Liu and Z. Zhen, Opt. Mater., 2015, 47, 256–262 CrossRef CAS .
- L. Zhong’An, W. Wenbo, L. Qianqian, Y. Gui, X. Li, L. Yunqi, Y. Cheng, Q. Jingui and L. Zhen, Angew. Chem., Int. Ed., 2010, 49, 2763–2767 CrossRef PubMed .
- H. Xu, M. Fu, S. Bo and X. Liu, RSC Adv., 2016, 6, 25023–25027 RSC .
- M. Malkoch, K. Schleicher, E. Drockenmuller, C. J. Hawker, T. P. Russell, P. Wu and V. V. Fokin, Macromolecules, 2005, 38, 3663–3678 CrossRef CAS .
- G. Deng, S. Bo, T. Zhou, H. Huang, J. Wu, J. Liu, X. Liu, Z. Zhen and L. Qiu, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 2841–2849 CrossRef CAS .
- E. E. Jelley, Nature, 1936, 138, 1009–1010 CrossRef CAS .
- O. Worz and G. Scheibe, Z. Naturforsch. B., 1969, 24, 381–390 CAS .
- B. Nina, B. L. Di and P. Gennaro, Chem. Soc. Rev., 2007, 36, 914–931 RSC .
- P. Lekha and E. Prasad, Chem.–Eur. J., 2010, 16, 3699–3706 CrossRef CAS PubMed .
- F. Ciardelli, G. Ruggeri and A. Pucci, Chem. Soc. Rev., 2012, 42, 857–870 RSC .
- B. Elissa, K. Christoph, K. Akshay, Z. Sven and W. Christoph, J. Appl. Polym. Sci., 2002, 86, 1235–1239 CrossRef .
- X. H. Zhou, J. Luo, S. Huang, T. D. Kim, Z. Shi, Y. J. Cheng, S. H. Jang, D. B. Knorr, R. M. Overney and A. K. Y. Jen, Adv. Mater., 2009, 21, 1976–1981 CrossRef CAS .
- R. R. Barto, C. W. Frank, P. V. Bedworth, E. Susan and R. E. Taylor, J. Chem. Phys., 2005, 122, 460–470 CrossRef PubMed .
- C. Weder, P. Neuenschwander, U. W. Suter, P. Pretre, P. Kaatz and P. Guenter, Macromolecules, 2002, 28, 2377–2382 CrossRef .
- G. Pescitelli, L. Di Bari and N. Berova, Chem. Soc. Rev., 2014, 43, 5211–5233 RSC .
- L. I. Markova, V. L. Malinovskii, L. D. Patsenker and R. Haner, Chem. Commun., 2013, 49, 5298–5300 RSC .
- A.-C. Le Duff, V. Ricci, T. Pliska, M. Canva, G. I. Stegeman, K. P. Chan and R. Twieg, Appl. Opt., 2000, 39, 947–953 CrossRef CAS PubMed .
- R. R. Barto, C. W. Frank, P. V. Bedworth, S. Ermer and R. E. Taylor, J. Chem. Phys., 2005, 122, 234907 CrossRef PubMed .
- V. I. Gavrilenko and M. A. Noginov, J. Chem. Phys., 2006, 124, 146–151 CrossRef PubMed .
- M. Frisch, G. Trucks, H. B. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. Petersson, Gaussian, Inc., Wallingford, CT, 2009, p. 200 Search PubMed .
- M. Q. He, T. M. Leslie and J. A. Sinicropi, Chem. Mater., 2002, 14, 2393–2400 CrossRef CAS .
Footnote |
| † Electronic supplementary information (ESI) available: Details of synthetic procedures, UV-vis spectra, thermoanalysis data and EO activity stability. See DOI: 10.1039/c6ra05320c |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.