
Influence of a surface modified Li anode on the electrochemical performance of Li–S batteries
Abstract
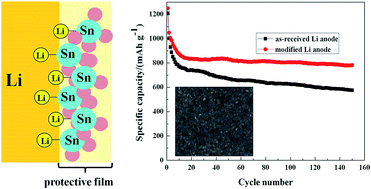
A bifunctional protective film is successfully prepared on Li metal surface by hexamethylditin modification in order to migrate the corrosion of sulfur-containing species and other electrolyte components. XRD, XPS, CV, SEM, AC impedance, polarization measurements and galvanostatic charge/discharge cycling tests are applied to characterize the film. The experimental results show that the protective film is dense and homogenous with a thickness of ∼10 nm. And under the protection of the film, the modified Li anode exhibits higher stability, lower interface resistance and a lower polarization potential difference than those of the as-received Li anode. Moreover, the cycling performance and rate capability of a Li/S cell are greatly improved by surface modification of the Li anode, delivering a discharge capacity of ∼800 mA h g−1 and a coulombic efficiency of more than 98.5% over 150 cycles at 0.2C as well as a notable rate performance with 510 mA h g−1 at 5C. After cycling, fewer sulfur-containing species are found on the modified Li anode surface than on the as-received Li anode surface, indicating the effective protection of the Li anode.
 Please wait while we load your content...
Please wait while we load your content...

