
A new approach to the determination of folic acid at trace levels: using a Fe(iii)-folic acid complex to amplify analytical signal
Abstract
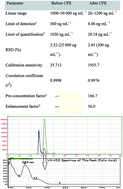
A fast, efficient, cost-effective, and environmental-friendly analytical methodology was developed for preconcentration and determination of trace folic acid in food samples prior to high performance liquid chromatography with diode array detection (HPLC-DAD). The method is based on formation of stable complexes between folic acid and Fe(III) ions at pH 8.0. The formed complexes were extracted to a nonionic surfactant phase containing PONPE 7.5. The surfactant rich phase (SRP) was separated by decantation and diluted with 300 μL of a mixture of 1 M HCl and methanol at a 1 : 1 ratio. The parameters and variables that affected the method were also investigated and optimized in detail. The limit of detection (LOD) of folic acid was 6.06 ng mL−1, the linear range of quantitation for folic acid was 20–1200 ng mL−1 and the correlation coefficients of the calibration curves were 0.9976. The average recoveries and relative standard deviations in the analysis of real samples were in the range of 95.1–105.1% and 1.73–5.25%, respectively. After validation of the method was carried out, the method was applied to the determination of folic acid in real samples, including baby foods, vegetables, cereals, and pharmaceutical samples.
 Please wait while we load your content...
Please wait while we load your content...

