
Exploring coumarin potentialities: development of new enzymatic inhibitors based on the 6-methyl-3-carboxamidocoumarin scaffold†
A. Fonsecaab,
M. J. Matosa,
J. Reisa,
Y. Duartec,
M. Gutiérrezc,
L. Santanab,
E. Uriarteb and
F. Borges*a
aCIQUP/Departamento de Química e Bioquímica, Faculdade de Ciências, Universidade do Porto, 4169-007, Porto, Portugal
bDepartamento de Química Orgánica, Faculdade de Farmacia, Universidade de Santiago de Compostela, 15782, Santiago de Compostela, Spain
cLaboratorio de Síntesis Orgánica, Instituto de Química de Recursos Naturales Universidad de Talca, Casilla 747, 3460000, Talca, Chile
First published on 12th May 2016
Abstract
Novel 6-methyl-3-carboxamidocoumarins (compounds 4–15) were synthesized by an effective three step synthetic strategy and screened towards MAO, AChE and BuChE enzymes. In general, the compounds act as selective MAO-B inhibitors. Compound 11 is highlighted as a potent (IC50 hMAO-B = 4.66 nM), reversible and non-competitive MAO-B inhibitor.
The increase in average life expectancy in developed countries has led to a rise in diagnosed cases of neurodegenerative diseases (ND's), namely Parkinson's (PD) and Alzheimer's (AD) diseases,1,2 and dementia. Currently none of these illnesses have an effective treatment to modify or stop their progress. The drugs currently available are only useful in delaying the progress of the diseases by controlling their symptoms.2,3 Monoamine oxidases (MAOs) are enzymes present in the outer mitochondrial membrane, which have two isoforms named MAO-A and MAO-B, that catalyze the oxidation of biogenic amines.4 Neurotransmitters, such as adrenaline, noradrenaline, dopamine, serotonin and β-phenylethylamine, are the main MAO substrates. Under normal conditions noradrenaline and serotonin are substrates of MAO-A while dopamine, a neurotransmitter present in low concentrations in the PD patient's brain, has a greater affinity for MAO-B.5 Activity of MAO-B is also linked to the production of reactive oxygen species (ROS) that cause oxidative stress and neuronal damage. Expression levels of MAO-B in neuronal tissue augment 4-fold with aging, resulting in an increase of dopamine metabolism and, therefore, higher production of hydrogen peroxide (H2O2).6 Thus, MAO-B inhibitors play an important role not only in dopamine metabolism but also in the reduction of brain oxidative damage. The involvement of MAO-B in AD is supported by the fact that neurons are extremely sensitive to oxidative stress as a consequence of: (a) their low content in endogenous antioxidants, such as glutathione, (b) the high proportion of an easily oxidized membrane covered by polyunsaturated fatty acids (c) the great oxygen brain consumption and also (d) a high content in iron.7–9 In addition, concerning AD, MAO-B activity and the coproduction of H2O2 and other type of ROS are also increased, leading to an amplification of the neuron oxidative stress damage process. The current therapy for PD is only palliative and is focused in curtailing the motor symptoms by restoring the dopamine levels, namely by the administration of L-dopa, a dopamine precursor, alongside with other drug co-adjuvants, such as dopamine agonists, catechol-o-methyltransferase (COMT) and MAO-B inhibitors, such as selegiline. For AD, the therapy is only focused on the administration of acetylcholinesterase (AChE) inhibitors that target the cholinergic system, considering that the disease is characterized by a cholinergic neuronal loss, and consequently acetylcholine (ACh) depletion.10 In brain synapses, ACh is hydrolyzed by AChE into choline and acetate.11 At present butyrylcholinesterase (BuChE) was also proposed as a druggable target and as a result both enzymes represent putative therapeutic targets for improving the cholinergic deficit responsible for the decline in cognitive, behavioral and global functioning characteristic of AD.12,13 Like in PD, none of the current drugs in therapy are able to modify disease progression, a condition that is well thought-out to be a driving force behind the ongoing research related to the discovery of new and potent inhibitors based on different types of scaffolds.14
Coumarins are heterocycles widely found in plants and other natural products that have synthetic accessibility and display remarkable biological properties, such as anticancer, antiviral, anti-inflammatory, antimicrobial and antioxidant agents.15–29 Previous studies have shown that coumarin is a noteworthy scaffold for the discovery and development of new potent and selective MAO-B and AChE inhibitors.25
Coumarins previously developed by our group (Fig. 1, structure A) have shown to display a remarkable potency and selectivity towards MAO-B activity. Till now our best-in-class IMAO-B coumarin was 3-(3-bromophenyl)-6-methylcoumarin (IC50 hMAO-B = 134 pM).30 The data attained so far stimulate the progress of the project and in accordance a lead optimization process was implemented in which the effect of a linker, located between the coumarin core and the exocyclic aromatic ring, was studied regarding IMAO activity. In addition, and taking advantage of the expenditure of the project it was also decided to move on from one-target to a dual-target drug design approach. So, other targets of interest in neurodegenerative diseases, like AChE, have been involved. The first studies were focused on the role of carbonylamine type linker (Fig. 1, structure B). From the study, potent and selective IMAO-B were attained which were also able to inhibit AChE in the range from 12 μM to 69 μM.29 The best dual candidate of the series was 3-(4′-chlorobenzamide)coumarin (IC50 hMAO-B = 1.95 μM and IC50 AChE = 18.71 μM). The best IMAO-B of the series was 3-(4′-methylbenzamide)-6-methylcoumarin (IC50 hMAO-B = 170 nM), which did not have relevant AChE inhibitory activity.
 | ||
| Fig. 1 Rational design followed in the present study to obtain the 3-substituted coumarins 4–15. | ||
So, additional studies focused in the effect of a carboxamide linker, located between coumarin and the exocyclic aromatic substituent, were accomplished. Within this framework new 6-methyl-3-carboxamidocoumarins (compounds 4–15, Fig. 1) were designed, synthesized and studied as MAO enzymatic inhibitors.
Coumarin derivatives (4–15) have been obtained efficiently by a three step synthetic strategy described in Scheme 1 and explained in detail in ESI.† Briefly, in the first step the coumarin used as starting material (compound 2) was synthesized by a Knoevenagel condensation, in which 5-methylsalicylaldehyde (1) was refluxed with diethyl malonate in ethanol, in presence of catalytic amounts of piperidine. After subsequent hydrolysis, compound 3 was obtained with an overall yield of 89%.31 Then, compounds 4–15 were synthesized by an amidation reaction in which the carboxylic acid 3 was activated with a coupling agent 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) in the presence of a nucleophilic catalyst 4-dimethylaminopyridine (DMAP).32 After adding the primary aromatic amine with the desired substitution pattern, 6-methyl-3-carboxamidocoumarin derivatives (4–15) have been obtained with yields ranging from 56% to 83%. Structural characterization of the compounds was performed by 1H and 13C NMR spectroscopy, mass spectrometry (EI-MS) and elemental analysis and is included in ESI.† The biological evaluation of the compounds 4–15 towards hMAO-A and hMAO-B was investigated by measuring their effects on the production of H2O2 from p-tyramine (a MAO substrate), using the Amplex Red MAO assay kit and recombinant hMAO with selegiline as reference compound.33 In addition, the inhibitory activities of the compounds 4–15 were evaluated towards Electrophorus electricus AChE and bovine serum BuChE using Ellman spectrophotometric method and galantamine as reference compound.34 The biological activity results expressed as IC50 values are listed in Table 1.
 | ||
| Scheme 1 Synthesis of coumarins 4–15. Reagents and conditions: (a) diethyl malonate, EtOH, piperidine, reflux, overnight. (b) NaOH (0.5% aq./EtOH), reflux, 4 h. (c) EDC, DMAP, DCM, corresponding amine, 0 °C to r.t., 4 h. | ||
| Compound | IC50 (nM) hMAO-A | IC50 (nM) hMAO-B | SI | IC50 (μM) AChE |
|---|---|---|---|---|
| a Inactive at 10 μM (highest concentration tested).b Values obtained under the assumption that the corresponding IC50 against MAO-A is the highest concentration tested (10 μM).c Inactive at 1000 μM (highest concentration tested).d Not determined. | ||||
| 4 | a | 11.80 ± 1.10 | >847.4b | c |
| 5 | a | 7.52 ± 1.05 | >1329.8b | 535.24 ± 0.01 |
| 6 | a | 13.90 ± 1.30 | >719.4b | 657.22 ± 0.01 |
| 7 | a | 160.60 ± 1.10 | >62.3b | 494.45 ± 0.03 |
| 8 | a | 10.10 ± 1.20 | >990.0b | 470.52 ± 0.18 |
| 9 | a | 296.90 ± 5.90 | >33.7b | c |
| 10 | a | 13.50 ± 1.10 | >740.7b | 621.23 ± 0.07 |
| 11 | a | 4.66 ± 1.13 | >2145.9b | 591.44 ± 0.02 |
| 12 | a | 11.40 ± 1.20 | >877.2b | 358.88 ± 0.05 |
| 13 | a | 18.30 ± 1.60 | >546.4b | 666.37 ± 0.11 |
| 14 | a | 45.40 ± 1.30 | >220.3b | c |
| 15 | a | 621.70 ± 1.80 | >16.1b | c |
| Selegiline | 68![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 730 ± 420 730 ± 420 |
17.00 ± 1.90 (ref. 3) | 4042.9 | d |
| Galantamine | d | d | d | 0.54 ± 0.50 |
In general, compounds 4–15 display a remarkable selectivity towards hMAO-B, as they were inactive against hMAO-A at the highest concentration tested, and an interesting structure-dependent inhibitory potency. The methyl (4–6) and bromine (10–12) derivatives, bearing substituents located at ortho, meta and para positions of the exocyclic aromatic ring, exhibit MAO-B activity in the low nanomolar range. In the case of the methoxy substituted coumarins (compounds 7–9), only the meta-substituted derivative display potency in the same range. For the hydroxy coumarin derivatives (compounds 13–15), it can be concluded that the MAO-B inhibitory activity is strongly dependent on the substituent location, being enhanced when they are located at ortho and meta positions. In summary, it was observed that the presence of electron donor substituents in the para position of the aryl ring attached to the amide group lead to a potency decrease, whereas derivatives bearing weak electron donors or acceptors do not change IMAO-B potency independently of their position. The 6-methyl-3-carboxamidocoumarins substituted in the meta position (compounds 5, 8 and 11) have a superior activity towards MAO-B than their ortho (compounds 4, 7 and 10) and para (compounds 6, 9 and 12) counterparts. In particular, compounds 5 (IC50 hMAO-B = 7.52 nM) and 11 (IC50 hMAO-B = 4.66 nM) showed hMAO-B inhibition at a low nanomolar range, slightly better than selegiline, and also benefiting from an excellent selective profile.
To examine the type of inhibition mechanism of the most promising hMAO-B inhibitor (compound 11) kinetic experiments were performed. For this purpose, the initial rates of the MAO-B-catalyzed oxidation of p-tyramine at five different substrate concentrations, in the absence or presence of the selected coumarin inhibitor, at different concentrations, were measured. The results are depicted in Fig. 2.
 | ||
| Fig. 2 Kinetic study on the mechanism of hMAO-B inhibition by compound 11. The effect of the inhibitors on the enzyme was determined from the double reciprocal plot of 1/rate (1/V) versus 1/substrate concentration in presence of varying concentrations of the inhibitors. The Ki value was calculated by the intersection of the curves obtained by plotting 1/V versus the inhibitor concentration for each substrate concentration (Dixon plots insets on the top right). | ||
Graphical analyses of the reciprocal Lineweaver–Burk plots allow the determination of Michaelis–Menten reaction kinetic parameters (Michaelis constant, Km and maximum velocity, Vmax). Concerning compound 11, it was found that the Km remained almost constant at different concentrations of the inhibitor whereas Vmax decreased. The Lineweaver–Burk plots obtained for different concentrations of compound 11 (Fig. 2) displayed a series of converging lines on the same point of the x-axis (1/[S]) profiling a non-competitive inhibition mechanism. From the Dixon plots, obtained from the replots of the slopes of the Lineweaver–Burk plots vs. inhibitor concentrations (upper right corner), the hMAO-B inhibition binding affinities, determined as inhibition constants (Ki), were calculated. As a result, compound 11 (Fig. 2) displayed a Ki value of 2.70 nM. The estimated Ki value correlated well with the inhibition mechanism suggested by the kinetic experiments, with the compound displaying IC50 and Ki values slightly different but within the low nanomolar range.
The reversibility of MAO-B inhibition by the test compound 11 was then assessed by time-dependent inhibition studies. The behavior of standard irreversible (R-(−)-deprenyl) and reversible (safinamide) inhibitors was also evaluated under the same experimental conditions. MAO-B activity (% of control) was measured along 60 minutes incubation with the enzyme inhibitors (Fig. 3). The analysis of time-dependent enzyme inhibition studies performed with the irreversible inhibitor (R-(−)-deprenyl, Fig. 3) showed that the enzyme residual activity decayed continuously after the first 15 minutes of incubation, which is consistent with irreversible enzymatic inhibition. In case of the reversible inhibitor safinamide (Fig. 3), an enhancement on enzymatic activity was observed along the analysis time. A similar behavior was observed for compound 11, which shows the gradual link to the allosteric binding site (non-competitive inhibitor) in the first 15 minutes, proceeded by the enhancement of enzymatic activity along the last 60 minutes, as its expectable of a MAO-B reversible inhibition profile.
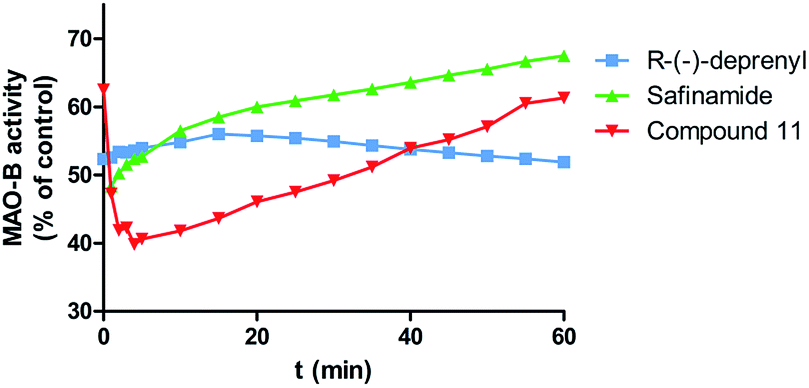 | ||
| Fig. 3 Time-dependent inhibition of recombinant human MAO-B by standard compounds (R)-(−)-deprenyl (50 nM), safinamide (40 nM) and test compound 11 (15 nM), the remaining activity was expressed as % of activity. Data are the mean ± S.D. of three different experiments. | ||
Finally, the drug-like properties of the compounds 4–15, namely the lipophilicity (expressed as the octanol/water partition coefficient, and herein called clogP) and other properties (molecular weight, number of hydrogen acceptors and donors and volume) were calculated using the Molinspiration calculation software. Topological polar surface area (TPSA) that has been shown to be a very good descriptor of drug absorption, including intestinal absorption, bioavailability, Caco-2 permeability and blood–brain barrier penetration was also calculated. The data are presented in Table 2. Complete procedures of in vitro biological studies, statistical analysis and drug-like properties calculations are listed in the ESI.†
| Compound | Molecular weight | clogP |
TPSA (Å2) | H-bond donor | H-bond acceptor | Volume (Å3) |
|---|---|---|---|---|---|---|
| 4/5/6 | 293.3 | 3.66/3.68/3.71 | 59.31 | 4 | 1 | 264.5 |
| 7/8/9 | 309.3 | 3.27/3.29/3.31 | 68.54 | 5 | 1 | 273.5 |
| 10/11/12 | 358.2 | 4.02/4.04/4.07 | 59.31 | 4 | 1 | 265.8 |
| 13/14/15 | 295.3 | 2.99/2.75/2.78 | 79.54 | 5 | 2 | 256.0 |
| Selegiline | 187.3 | 2.64 | 3.24 | 1 | 0 | 202.6 |
| Galantamine | 287.4 | 1.54 | 41.93 | 4 | 1 | 268.2 |
Analyzing the results for the inhibitory activity towards human AChE depicted in Table 1, one can conclude that all compounds presented a moderate inhibitory activity (micromolar range) towards the enzyme. None of the tested compounds displayed a noticeable activity towards BuChE at the highest concentration tested (10 mM) (data not shown). Compound 12, the para-bromine coumarin derivative, was found to be the most active compound towards AChE (IC50 = 358.88 μM). Analysing the overall data, one can conclude that the 6-methyl-3-carboxamidocoumarins substituted with a hydroxyl substituent (compounds 13–15) are the less potent compounds of the series. However, when a methyl or methoxy substituent (compounds 5 and 8) is located at meta position of the aromatic exocyclic ring a slight increment of inhibitory activity is observed, when compared with their ortho (compounds 4 and 7) and para (compounds 6 and 9) counterparts.
In our previous study, compound 6 analogue (compound 4 in ref. 29), which also have a methyl substituent, in para position, had displayed a IC50 hMAO-B = 170 nM, which is ten times lower. Nevertheless, it showed a superior affinity to AChE than the compounds presented here.29 Thus, it can be concluded that the carboxamide spacer, and specially the location of the carbonyl group, is a key feature for MAO-B and AChE inhibitory activities.
Additionally, from the prediction drug-like properties of compounds 4–15 (Table 2) it can be observed that no violations of Lipinski's rule (molecular weight, logP, number of hydrogen donors and acceptors) were found and that the TPSA, described as a predictive indicator of the drug capacity of membrane penetration, is favorable. Therefore, the data provided a preliminary indication that this type of compounds can cross membranes and act in the central nervous system.
The remarkable results found for compounds 5 and 11 (hMAO-B IC50 of 7.52 and 4.66 nM respectively) encourage us to continue our research based on the coumarin scaffold. Compound 11 acts as a potent, selective, reversible and non-competitive MAO-B inhibitor. In addition, compound 12 (hMAO-B IC50 of 11.40 nM and AChE IC50 of 358.88 μM) can be looked as a stimulating framework to develop dual target MAO-B/AChE inhibitors. Further examination of the cytotoxic and pharmacokinetic properties of compounds 11 and 12 is important to define which one will be a candidate for in vivo studies. In summary, the data attained so far is a noteworthy contribution for the development of new drug candidates for PD and AD based on 6-methylcoumarin scaffold.
Acknowledgements
The authors would like to thank Fundação para a Ciência e Tecnologia (FCT) – QUI/UI0081/2015-POCI-01-0145-FEDER-006980 for the financial support. Thanks are due to FCT, POPH and QREN for the post-doctoral and doctoral grants: A. Fonseca (SFRH/BD/80831/2011) and J. Reis (SFRH/BD/96033/2013) and M. J. Matos (SFRH/BPD/95345/2013).Notes and references
- R. J. Castellani, R. K. Rolston and M. a. Smith, Dis.-Mon., 2010, 56, 484–546 CrossRef PubMed.
- L. M. L. de Lau and M. M. B. Breteler, Lancet Neurol., 2006, 5, 525–535 CrossRef PubMed.
- M. R. Farlow and J. L. Cummings, Am. J. Med., 2007, 120, 388–397 CrossRef CAS PubMed.
- J. P. Johnson, Biochem. Pharmacol., 1968, 17, 1285–1297 CrossRef.
- M. B. Youdim, D. Edmondson and K. F. Tipton, Nat. Rev. Neurosci., 2006, 7, 295–309 CrossRef CAS PubMed.
- A. Gaspar, N. Milhazes, L. Santana, E. Uriarte, F. Borges and M. J. Matos, Curr. Top. Med. Chem., 2015, 15, 432–445 CrossRef CAS PubMed.
- P. Riederer, Neurotoxicology, 2004, 25, 271–277 CrossRef CAS PubMed.
- V. Jain, M. C. Langham and F. W. Wehrli, J. Cereb. Blood Flow Metab., 2010, 30, 1598–1607 CrossRef CAS PubMed.
- T. A. Rouault, Nat. Rev. Neurosci., 2013, 14, 551–564 CrossRef CAS PubMed.
- M. Itakura, H. Nakajima, T. Kubo, Y. Semi, S. Kume, S. Higashida, A. Kaneshige, M. Kuwamura, N. Harada, A. Kita, Y.-T. Azuma, R. Yamaji, T. Inui and T. Takeuchi, J. Biol. Chem., 2015, 290, 26072–26087 CrossRef CAS PubMed.
- V. N. Talesa, Mech. Ageing Dev., 2001, 122, 1961–1969 CrossRef CAS PubMed.
- M. Khoobi, M. Alipour, A. Moradi, A. Sakhteman, H. Nadri, S. F. Razavi, M. Ghandi, A. Foroumadi and A. Shafiee, Eur. J. Med. Chem., 2013, 68, 291–300 CrossRef CAS PubMed.
- R. M. Lane, S. G. Potkin and A. Enz, Int. J. Neuropsychopharmacol., 2006, 9, 101–124 CrossRef CAS PubMed.
- M. Singh, M. Kaur, H. Kukreja, R. Chugh, O. Silakari and D. Singh, Eur. J. Med. Chem., 2013, 70, 165–188 CrossRef CAS PubMed.
- F. Borges, F. Roleira, N. Milhazes, L. Santana and E. Uriarte, Curr. Med. Chem., 2005, 12, 887–916 CrossRef CAS PubMed.
- F. Borges, F. M. F. Roleira, N. Milhazes, E. Uriarte and L. Santana, Front. Med. Chem., 2009, 4, 23–85 Search PubMed.
- M. Riveiro, N. De Kimpe, A. Moglioni, R. Vazquez, F. Monczor, C. Shayo and C. Davio, Curr. Med. Chem., 2010, 17, 1325–1338 CrossRef CAS PubMed.
- M. J. Matos, S. Vazquez-Rodriguez, L. Santana, E. Uriarte, C. Fuentes-Edfuf, Y. Santos and A. Munoz-Crego, Med. Chem., 2012, 8, 1140–1145 CAS.
- D. Viña, M. J. Matos, G. Ferino, E. Cadoni, R. Laguna, F. Borges, E. Uriarte and L. Santana, ChemMedChem, 2012, 7, 464–470 CrossRef PubMed.
- M. J. Matos, S. Vazquez-Rodriguez, E. Uriarte, L. Santana and D. Viña, Bioorg. Med. Chem. Lett., 2011, 21, 4224–4227 CrossRef CAS PubMed.
- D. Secci, S. Carradori, A. Bolasco, P. Chimenti, M. Yáñez, F. Ortuso and S. Alcaro, Eur. J. Med. Chem., 2011, 46, 4846–4852 CrossRef CAS PubMed.
- S. Vazquez-Rodriguez, M. J. Matos, L. Santana, E. Uriarte, F. Borges, S. Kachler and K. N. Klotz, J. Pharm. Pharmacol., 2013, 65, 697–703 CrossRef CAS PubMed.
- I. Kostova, S. Bhatia, P. Grigorov, S. Balkansky, V. S. Parmar, A. K. Prasad and L. Saso, Curr. Med. Chem., 2011, 18, 3929–3951 CrossRef CAS PubMed.
- M. J. Matos, P. Janeiro, R. M. González Franco, S. Vilar, N. P. Tatonetti, L. Santana, E. Uriarte, F. Borges, J. A. Fontenla and D. Viña, Future Med. Chem., 2014, 6, 371–383 CrossRef CAS PubMed.
- M. J. Matos, D. Viña, E. Quezada, C. Picciau, G. Delogu, F. Orallo, L. Santana and E. Uriarte, Bioorg. Med. Chem. Lett., 2009, 19, 3268–3270 CrossRef CAS PubMed.
- M. J. Matos, D. Viña, C. Picciau, F. Orallo, L. Santana and E. Uriarte, Bioorg. Med. Chem. Lett., 2009, 19, 5053–5055 CrossRef CAS PubMed.
- M. J. Matos, S. Vazquez-Rodriguez, L. Santana, E. Uriarte, C. Fuentes-Edfuf, Y. Santos and A. Muñoz-Crego, Molecules, 2013, 18, 1394–1404 CrossRef CAS PubMed.
- M. J. Matos, C. Terán, Y. Pérez-Castillo, E. Uriarte, L. Santana and D. Viña, J. Med. Chem., 2011, 54, 7127–7137 CrossRef CAS PubMed.
- D. Viña, M. J. Matos, M. Yáñez, L. Santana and E. Uriarte, Med. Chem. Commun., 2012, 3, 213–218 RSC.
- M. J. Matos, S. Vilar, V. Garcia-Morales, N. P. Tatonetti, E. Uriarte, L. Santana and D. Viña, ChemMedChem, 2014, 9, 1488–1500 CrossRef CAS PubMed.
- F. Chimenti, B. Bizzarri, A. Bolasco, D. Secci, P. Chimenti, A. Granese, S. Carradori, D. Rivanera, A. Zicari, M. Scaltrito and F. Sisto, Bioorg. Med. Chem. Lett., 2010, 20, 4922–4926 CrossRef CAS PubMed.
- C. Murata, T. Masuda, Y. Kamochi, K. Todoroki, H. Yoshida, H. Nohta, M. Yamaguchi and A. Takadate, Chem. Pharm. Bull., 2005, 53, 750–758 CrossRef CAS PubMed.
- M. Yáñez, N. Fraiz, E. Cano and F. Orallo, Biochem. Biophys. Res. Commun., 2006, 344, 688–695 CrossRef PubMed.
- P. Torre, L. Saavedra, J. Caballero, J. Quiroga, J. Alzate-Morales, M. Cabrera and J. Trilleras, Molecules, 2012, 17, 12072–12085 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra05262b |
| This journal is © The Royal Society of Chemistry 2016 |