
A ferrocenyl pyridine-based Ru(II) arene complex capable of generating ·OH and 1O2 along with photoinduced ligand dissociation†
Tianji Wangab,
Qianxiong Zhou*a,
Yangyang Zhangab,
Yue Zhengab,
Weibo Wanga,
Yuanjun Houa,
Guoyu Jianga,
Xuexin Chenga and
Xuesong Wang*a
aKey Laboratory of Photochemical Conversion and Optoelectronic Materials, Technical Institute of Physics and Chemistry, Chinese Academy of Sciences, Beijing 100190, P. R. China. E-mail: xswang@mail.ipc.ac.cn; zhouqianxiong@mail.ipc.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, P. R. China
First published on 27th April 2016
Abstract
Photoactivated chemotherapy (PACT) and photodynamic therapy (PDT) are two types of cancer treatment modalities that rely on photoinduced ligand dissociation and photosensitized or photocatalyzed reactive oxygen species (ROS) generation to realize anticancer activities with high selectivity, respectively. In this study, a novel Ru complex, [(p-cym)Ru(bpy)(py-Fc)]2+ (1), where p-cym = para-cymene, bpy = 2,2′-bipyridine, and py-Fc = 4-pyridyl ferrocene, was demonstrated to show dual activity from PACT and PDT. 1 has a long-wavelength absorption band extending to 600 nm and can generate both hydroxyl radicals (·OH) and singlet oxygen (1O2) along with photoinduced monodentate ligand dissociation. As a result, 1 displays DNA photocleavage activity as well as phototoxicity against human ovarian adenocarcinoma cells SKOV3 and human lung adenocarcinoma cells A549. The underlying mechanisms were discussed by comparing the photophysical, photochemical, and photobiological properties of 1 with that of [(p-cym)Ru(bpy)(py)]2+ (2, py = pyridine) and free py-Fc, providing guidelines for developing new photoactivated metallodrugs with multiple functions.
Introduction
The worldwide clinical applications of cisplatin, carboplatin, and oxaliplatin make transition metal complexes the most promising candidates for new anticancer agents.1 Main concerns for the use of these drugs are the side effects of current Pt-based metallodrugs that are generally believed to be the result of their poor tumor selectivity. To address this issue, photoactivated chemotherapy (PACT) is drawing increasing attention as it may localize drug activity in diseased tissues by spatial and temporal control of irradiation.2 In this context, the photoinduced ligand dissociation reaction has found intriguing applications in the development of PACT agents. One prominent example is trans, trans, trans-[Pt(N3)2(OH)2(NH3)(py)] (py = pyridine), which is stable in the dark and non-toxic, but becomes 80-fold more toxic toward cancer cells than cisplatin after photoinduced ligand dissociation and reduction reactions.3Similar to cisplatin, many Ru(II) complexes with photo-labile ligand(s) can bind DNA covalently after photoinduced ligand dissociation and therefore, have been studied intensively and extensively as new types of PACT agents.4 When the photo-labile ligand has an anticancer capability on its own, better efficacy is expected as the result of the synergistic effect.5 More interestingly, some novel Ru(II) complexes have been constructed to generate singlet oxygen (1O2) along with photoinduced ligand dissociation resulting from the lowest-lying ligand-centered 3ππ* state and the high-lying metal-centered ligand-field (3LF) state, respectively.6 Very recently, we found that [Ru(bpy)2(py-SO3)]+ and its derivatives (bpy = 2,2′-bipyridine and py-SO3 = pyridine-2-sulfonate) were able to undergo Ru–O homolysis upon irradiation, leading to the dissociation of py-SO3 from the Ru center in a radical manner that may further produce hydroxyl radicals (·OH) by reactions with water.7 Both 1O2 and ·OH are reactive oxygen species (ROS) responsible for photodynamic therapy (PDT), another type of photoactivation based cancer treatment modality that has clinical applications,8 rendering the corresponding complexes to possess dual activity from PACT and PDT.
In this paper, we explored a new strategy to combine PACT with PDT in a Ru(II) arene complex. Ru(II) arene complexes of the type [(η6-arene)Ru(X)(Y)(Z)]n+, in which X is a monodentate ligand (usually halide), Y and Z are monodentate or chelating ligands, show unique anticancer activities, in particular, toward cisplatin-resistant cancers and metastatic tumors.9 A crucial step in one mode of activation of these anticancer agents is usually the initial aquation of the Ru–X bond to form a more reactive species.10 By delicate combinations of the monodentate and bidentate ligands, Sadler and coworkers reported the first Ru(II) arene-based PACT agent, [(p-cym)Ru(bpm)(py)][PF6]2 (where p-cym = para-cymene, bpm = 2,2′-bipyrimidine).10 Using a BODIPY chromophore-modified pyridine as the photolabile ligand, we realized its photo-dissociation from [(η6-p-cym)Ru(bpy) (py-BODIPY)](PF6)2, with longer wavelength light. The efficient photo-dissociation is attributed to photoinduced electron transfer from the BODIPY chromophore to the Ru(II) arene moiety that weakens the coordination ability of py-BODIPY remarkably.11 In contrast, [(η6-p-cym)Ru(bpy)(py)](PF6)2 (complex 2 in Scheme 1) is stable even when irradiated with UV light.11
 | ||
| Scheme 1 Chemical structures of the complexes 1, 2 and py-Fc. | ||
To render Ru(II) arene complexes dually active to PACT and PDT, we herein examined the photochemical property of [(η6-p-cym)Ru(bpy)(py-Fc)](PF6)2 (complex 1 in Scheme 1, where py-Fc = 4-pyridyl ferrocene). The strong electron-donating capability of the ferrocenyl group may favor photoinduced intramolecular electron transfer in 1, leading to an efficient dissociation of the py-Fc ligand. More importantly, besides its potential therapeutic effect toward hypoferric anemia, ferrocene has been widely used as a unique pharmacophore to construct new anticancer agents.12 The in vitro and in vivo anti-proliferative activity of a ferrocenium salt was observed in as early as 1984.13 While the anticancer activities of many ferrocenyl derivatives are associated with transformation into the ferrocenium form, due to their low oxidation potentials accessible in physiological conditions, the generation of ·OH has proven to play a pivotal role in the mechanism of action of ferrocenium ions.14 Moreover, the released Fe2+/Fe3+ ions due to the decomposition of either ferrocenium or ferrocenyl groups may also catalyze ·OH generation.15 Bearing these considerations in mind, the photophysical, photochemical and photobiological properties of 1 were compared in detail with 2 and py-Fc. Interestingly, we found that 1 can not only generate ·OH, but also 1O2 along with photoinduced monodentate ligand dissociation. Agarose gel electrophoresis and MTT assays demonstrate that 1 may photo-cleave DNA and photo-inactivate human ovarian adenocarcinoma SKOV3 cells and human lung adenocarcinoma A549 cells effectively while py-Fc and 2 are inert. Though efforts to modify Ru-based chemotherapeutics with a ferrocenyl group to take advantage of this synergistic effect have been reported,16 to the best of our knowledge, 1 represents the first example to utilize the ferrocenyl modification to transform a stable ligand into a photolabile one while also rendering the complex able for ROS generation.
Results and discussion
Photophysical and electrochemical property
The UV-vis absorption spectra of 1, 2 and py-Fc (10 μM) in CH3OH are shown in Fig. 1. The parent, complex 2, displays two absorption peaks centered at 305 and 317 nm, respectively, and a low intensity absorption tail extending to 430 nm, assignable to the mixed 1LF–1MLCT transitions.10 In contrast, the free py-Fc ligand shows four absorption bands with maxima at 245, 281, 350 and 452 nm, respectively. Upon coordination to the Ru(II) center, all of these bands experience a remarkable red shift. In particular, the lowest energy absorption band of py-Fc has a bathochromic shift as large as 570 nm, making the absorption onset of 1 extended to 600 nm. Such a spectrum shift is favorable for both PACT and PDT applications because longer photoactivation wavelengths correspond to deeper tissue penetration. To understand the underlying mechanism of this red shift, we examined the protonation effect of py-Fc. As shown in Fig. S1,† addition of CF3COOH into the alcohol solution of py-Fc led to a similar red shift in the absorption spectrum. This result suggests that all absorption bands of py-Fc may have charge-transfer (CT) character directed from the Fc moiety to the py moiety. Thus, either protonation or coordination may facilitate the CT processes by strengthening the electron-withdrawing capability of the py group, accompanied by the narrowing of the CT transition energies. Similar to 2, 1 is not fluorescent, a common characteristic for most Ru(II) arene complexes.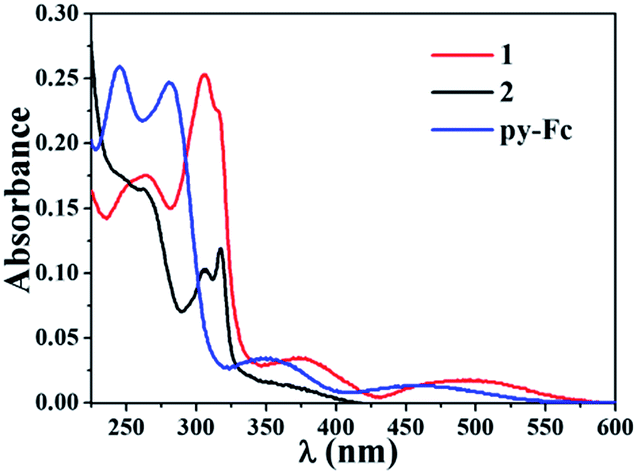 | ||
| Fig. 1 UV-vis absorption spectra of 1, 2 and py-Fc (10 μM) in CH3OH. | ||
As shown in Fig. S2† and Table 1, py-Fc displays the typical redox process of the Fc group at +0.56 V (vs. SCE). Upon cathodic scanning, a redox wave appears at −0.77 V, attributable to the reduction of the py moiety. For 2, a bpy-based reduction process occurs at −0.76 V. As a combination of 2 and py-Fc, 1 displays a Fc+/Fc-based wave at 0.56 V. The redox process occurred at −0.76 V may be attributed to the reduction of the bpy and py-Fc ligands.
Ligand photo-dissociation
An ideal PACT agent should be stable enough in the dark to keep nontoxic and active enough upon irradiation to release cytotoxic species. We first examined the ligand exchange activity of 1 by monitoring its UV-visible spectra changes. No discernible spectra changes were found upon standing in the dark for 24 h (Fig. S3†). Upon visible light irradiation (>400 nm), the lowest-energy absorption band of 1, resulting mainly from py-Fc, experienced a significant blue shift, hinting at its dissociation from the Ru(II) core (Fig. 2). Additionally, other spectrum changes, e.g. absorbance decrease between 300 and 330 nm and increase around 250 nm, were also in good agreement with the dissociation of py-Fc. | ||
| Fig. 2 UV-vis absorption spectra changes of 1 (10 μM) in CH3OH upon irradiation (>400 nm). | ||
1H NMR data can provide more direct evidence of the photo-dissociation of py-Fc. The chemical shifts of free py-Fc at 4.05 ppm, 4.89 ppm and 7.52 ppm may be assigned to the 5Hs of the unsubstituted cyclopentadiene, 2H on the 3′ and 4′-position of the substituted cyclopentadiene, and 2H on 3 and 5-position of py, respectively (Fig. 3). When coordinated onto Ru(II) core, they shift to 3.98 ppm, 4.85 ppm (buried in water peak in CD3OD) and 7.44 ppm, respectively. The chemical shift changes give us the opportunity to follow the photoinduced ligand exchange reaction of 1. As shown in Fig. 3, about half of the 1 molecules underwent monodentate ligand dissociation after 3 h of irradiation (>400 nm).
 | ||
| Fig. 3 1H NMR spectra changes of 1 upon irradiation (>400 nm) for 180 min. | ||
Unlike 1, no ligand dissociation was observed for 2 even under UV irradiation (>300 nm).11 The different photochemical properties of 1 and 2 may be attributed to the ferrocenyl modification. Due to the electron-donating feature of the ferrocenyl group, py-Fc is expected to coordinate metal more efficiently than py. Upon irradiation, however, the intramolecular electron transfer from py-Fc to Ru(II) arene, a thermodynamically allowed process (−0.75 eV) estimated from their redox potentials (0.56 V and −0.76 V) and excitation energy (2.07 eV corresponding to the absorption onset of 600 nm), may weaken the coordination ability of py-Fc markedly due to the emergence of a positive charge on it. No any transient absorption spectra were recorded, suggesting the electron transfer and charge recombination occurred in the time domain shorter than the response of our experimental setup (ns).
ROS generation
The anticancer activities of many ferrocenyl and ferrocenium compounds are highly associated with the generation of ·OH.14 This prompted us to evaluate ·OH activities of 1 and py-Fc. Using 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) as spin-trapping agent, we observed a four-line signal with intensity ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:2:1 and a hyperfine splitting constant of 14.9 G upon visible light irradiation (>400 nm) of 1 (Fig. 4) or py-Fc (Fig. S4†) in CH3CN/H2O (1:1), which may be assigned to the EPR signal of the adduct of DMPO and ·OH.16 The signal was quenched efficiently by KI (Fig. 4), a common scavenger of ·OH, confirming the assignment further. Control experiments showed that 2 did not generate ·OH even when exposed to UV light, suggesting the ·OH generation of 1 originates from py-Fc. Similar EPR signal intensities were obtained in the cases of 1 and py-Fc after 2 min irradiation, and the photoinduced ligand dissociation of 1 may be neglected during this period (see Fig. 2), which indicates that py-Fc retains its ·OH generation capability when coordinated on Ru(II) core. Because no ·OH signal could be detected in the dark, the ·OH generation of 1 and free py-Fc may result from their photoinduced electron transfer to oxygen. However, no DMPO-OOH adduct signals were observed, probably because the dismutation reaction rate of the superoxide anion radical (O2˙−) is as high as 2.4 × 105 M−1 s−1 at pH 7.4,17 and the reaction rate constant of DMPO with ·OH is higher than O2˙− by nearly 8 orders of magnitude.18
:2:2:1 and a hyperfine splitting constant of 14.9 G upon visible light irradiation (>400 nm) of 1 (Fig. 4) or py-Fc (Fig. S4†) in CH3CN/H2O (1:1), which may be assigned to the EPR signal of the adduct of DMPO and ·OH.16 The signal was quenched efficiently by KI (Fig. 4), a common scavenger of ·OH, confirming the assignment further. Control experiments showed that 2 did not generate ·OH even when exposed to UV light, suggesting the ·OH generation of 1 originates from py-Fc. Similar EPR signal intensities were obtained in the cases of 1 and py-Fc after 2 min irradiation, and the photoinduced ligand dissociation of 1 may be neglected during this period (see Fig. 2), which indicates that py-Fc retains its ·OH generation capability when coordinated on Ru(II) core. Because no ·OH signal could be detected in the dark, the ·OH generation of 1 and free py-Fc may result from their photoinduced electron transfer to oxygen. However, no DMPO-OOH adduct signals were observed, probably because the dismutation reaction rate of the superoxide anion radical (O2˙−) is as high as 2.4 × 105 M−1 s−1 at pH 7.4,17 and the reaction rate constant of DMPO with ·OH is higher than O2˙− by nearly 8 orders of magnitude.18
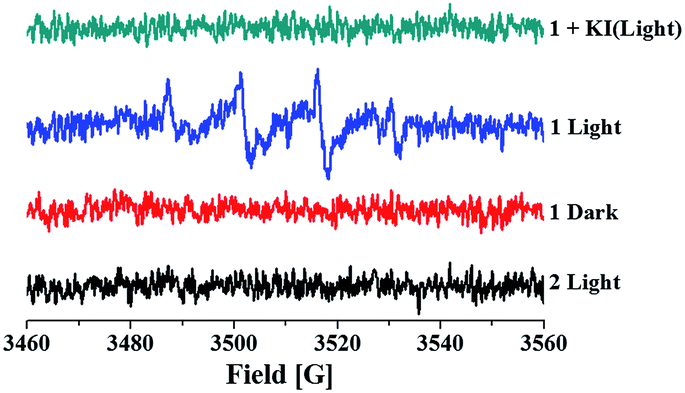 | ||
| Fig. 4 EPR signals of 1 or 2 (1 mM) and DMPO (50 mM) in air-saturated PBS/CH3CN (1:1) in the dark or upon irradiation (>400 nm) for 2 min. | ||
Surprisingly, and interestingly, we found that 1 is able to generate 1O2 as well. Upon visible light irradiation (>400 nm) of a solution of 1 and 2,2,6,6-tetramethyl-4-piperidone (TEMP) in air-saturated CH3CN, a three-line signal with a hyperfine coupling constant of 16.0 G appeared (Fig. 5), in line with the signal of TEMPO (the adduct of TEMP and 1O2).19 The signal was quenched once NaN3, a scavenger of 1O2, was added, vindicating the 1O2 assignment.
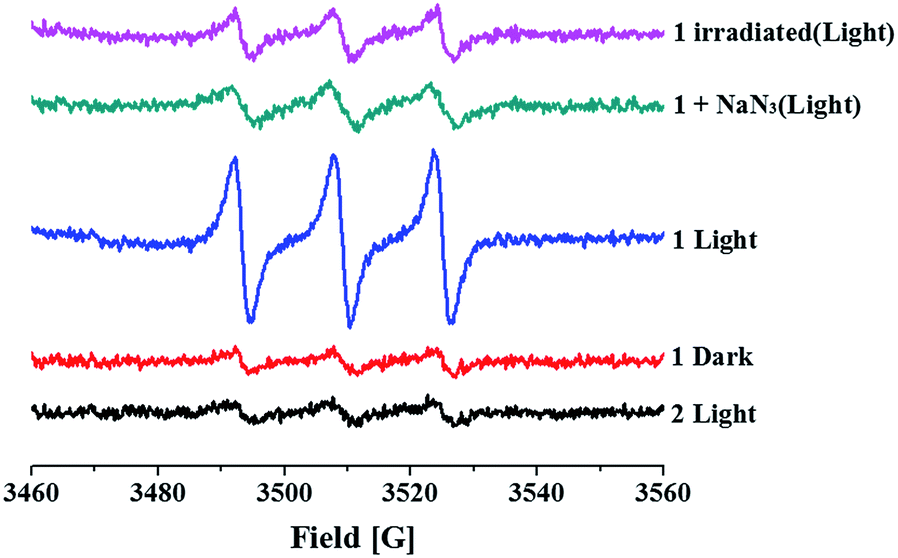 | ||
| Fig. 5 EPR signals of 1 or 2 (1 mM) and TEMP (50 mM) in air-saturated CH3CN in the dark or upon irradiation (>400 nm) for 2 min. 1 irradiated represents the sample that has been irradiated for 2 h prior to EPR measurement. | ||
In sharp contrast, no 1O2 generation was detected for both 2 (Fig. 5) and py-Fc (Fig. S5†) under the same conditions. Additionally, the 1O2 generation efficiency decreased dramatically after the solution of 1 was subjected to visible light irradiation (>400 nm) for 2 h (Fig. 5), suggesting that 1O2 originates from 1 rather than from free py-Fc dissociated from the Ru centre and the in situ formed Ru product. Obviously, the combination of 2 and py-Fc in a single molecule offers 1 a new ability to generate 1O2. In our previous work, the lowest-energy 3MLCT state of 2 was estimated to be 2.32 eV using TD-DFT (time dependent density functional theory).11 Assuming the lowest-energy triplet excited state of py-Fc (3py-Fc*) is similar to that of ferrocene (1.8 eV),20 a pathway accessible to 3py-Fc* may be established by ultra-efficient intersystem crossing (ISC) from 1MLCT to 3MLCT followed by internal conversion (IC) from 3MLCT to 3py-Fc*, from which 1O2 is generated by way of energy transfer. The short lifetime of the 3MLCT state of 2 and low intersystem crossing efficiency of free py-Fc may account for their inability towards 1O2 generation. Additionally, the heavy metal effect from the Ru atom may also play a role in promoting the access of 3py-Fc* directly from 1py-Fc*.
Chemical trapping methods were also utilized to characterize ROS generation by 1. Terephthalic acid (TA) can react with ·OH to form highly fluorescent 2-hydroxyterephthalic acid (HTA).21 Visible light irradiation of an aqueous solution of 1 and TA gave rise to a fluorescence enhancement as shown in Fig. 6. When KI was present in the irradiated solution, the fluorescence intensity of the solution remained a constant background value (Fig. S6†). These results further demonstrate the ·OH generation ability of 1.
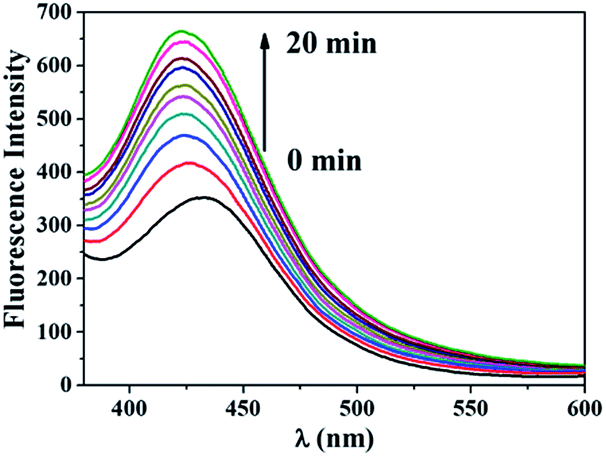 | ||
| Fig. 6 Fluorescence intensity changes of an aqueous solution containing 1 mM TA, 2 mM NaOH and 100 μM 1 upon irradiation at 470 nm (LED, 0.32 mW cm−2). | ||
To trap 1O2, fluorescent 1,3-diphenylisobenzofuran (DPBF) is an ideal reagent that can react with 1O2 efficiently to form a non-emissive product.22 Using [Ru(bpy)3]2+ as the standard (ΦΔ = 0.57 in CH3CN),23 the 1O2 quantum yield of 1 was measured to be 0.13, while the 1O2 generation ability of 2 and py-Fc may be neglected (Fig. 7). All these results are very consistent with EPR experiments.
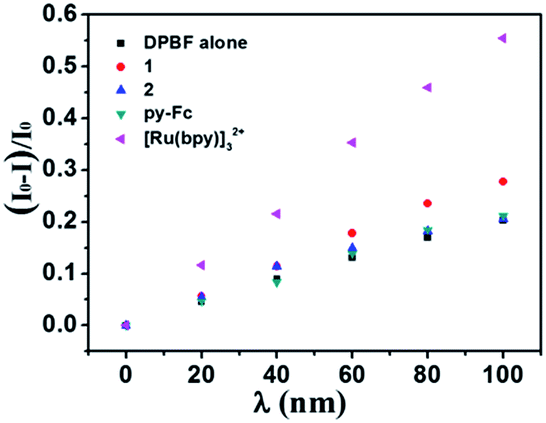 | ||
| Fig. 7 DPBF fluorescence bleaching (λem = 479 nm, λex = 440 nm) in air-saturated CH3CN upon irradiation at 380 nm (light source in spectrophotometer) in the presence of 1, 2, py-Fc or [Ru(bpy)3]2+. | ||
DNA photo-damage
The ligand photo-dissociation property and the ROS generation ability of 1 encouraged us to study its photo-damage potential toward DNA. As shown in Fig. 8, noticeable single strand cleavage, as evidenced by the transformation of pUC19 plasmid DNA from the supercoiled circular form (SC) to the nicked circular form (NC), was found after 15 min of visible light irradiation in the presence of 10 μM 1. Increasing the concentration of 1 led to improved DNA cleavage. In the absence of either irradiation or O2, 1 lost its DNA cleavage ability (Fig. 9, Lane 2, 8 and 10). Both KI and NaN3 restricted the DNA cleavage dramatically (Fig. 9, Lane 6 and 7), while catalase and SOD, the scavengers of H2O2 and O2˙− respectively, had no effect (Fig. 9, Lane 4 and 5). All findings indicate that ·OH and 1O2 are responsible for the DNA photo-cleavage ability of 1. Additionally, no DNA mobility retardation, resulting generally from covalent binding of metal complexes toward DNA, was observed in our experiments probably due to the short irradiation time and thus, less efficient ligand dissociation. | ||
| Fig. 8 Agarose gel electrophoresis pattern of supercoiled pUC19 plasmid DNA (40 μg mL−1) in air-saturated PBS (pH = 7.4, 5 mM) after irradiation (>400 nm) for 15 min in the presence of varied concentrations of 1. Lane 1, DNA alone; Lane 5, DNA alone in the dark; Lane 2–4 and 6–8, the concentrations of 1 are 1, 5, 10, 25, 50, and 100 μM, respectively. SC and NC represent supercoiled circular and nicked circular forms, respectively. | ||
 | ||
| Fig. 9 Agarose gel electrophoresis pattern of supercoiled pUC19 plasmid DNA (40 μg mL−1) in air-saturated PBS (pH = 7.4, 5 mM) upon irradiation (>400 nm) for 15 min in the presence of 1 (50 μM) and different additives. Lane 1 and 11, DNA alone; Lane 2 and 8, DNA + 1 (dark); Lane 3 and 9, DNA + 1; Lane 4, DNA + 1 + catalase (1000 U mL−1); Lane 5, DNA + 1 + SOD (1000 U mL−1); Lane 6, DNA + 1 + KI (50 mM); Lane 7, DNA + 1 + NaN3 (50 mM); Lane 10, DNA + 1 + Ar. SC and NC represent supercoiled circular and nicked circular forms, respectively. | ||
In sharp contrast, both 2 and py-Fc cannot photo-cleave DNA even at a high concentration of 100 μM (Fig. S7†). While the behavior of 2 is understandable, due to its disability to generate either ·OH or 1O2, that of py-Fc is really out of our expectation. The lack of electrostatic attraction between py-Fc and DNA may play a role, leading to a weak binding affinity of py-Fc to DNA, and thus, a poor bioavailability of ·OH generated by py-Fc. Additionally, the oil–water partition coefficient (P) of py-Fc is two orders of magnitude larger than that of 1 (Table 1), implying a high propensity of aggregation in aqueous solutions, which may greatly impair its binding to DNA and its ·OH generation ability.
Cytotoxicity
The ligand photo-dissociation and ROS generation activities of 1 may facilitate its application as an anticancer agent. To explore this potential, the cytotoxicity of 1 in the dark and under 470 nm irradiation (LED, 0.32 mW cm−2) were examined against human ovarian SKOV3 cells and human lung adenocarcinoma A549 cells. While the dark cytotoxicity was assayed by the MTT method after incubating the cells with 1, 2 or py-Fc for 24.5 h, the phototoxicity measurements were conducted following a procedure of 4 h incubation in the dark, then 0.5 h irradiation, and then 20 h incubation in the dark.As shown in Fig. 10, irradiation alone has no effect on the viability of both cell lines. 1 displayed remarkable photoactivated anticancer property. For example, the viability of SKOV3 and A549 cells declined to 30% and 38%, respectively, in the presence of 70 μM 1 and 0.5 h irradiation. In contrast, 87% and 76% cells were still alive at the same concentration of 1 in the dark. The phototoxicity of 1 is not yet satisfactory, presumably due to the weak irradiation intensity. Considering the weak irradiation intensity (corresponding to only 0.6 J cm−2 in our experiments) and the poor covalent binding ability of 1 toward DNA, as shown in Fig. 8, the cytotoxicity of 1 resulted most likely from ROS generation rather than from the in situ formed Ru fragments. Turro, Dunbar and coworkers compared the phototoxicities of [Ru(tpy)(CH3CN)3]2+ and [Ru(tpy)(5CNU)3]2+, where tpy = 2,2′:6′,2′′-terpyridine and 5CNU = 5-cyanouracil.4b While [Ru(tpy)(5CNU)3]2+ exhibited a toxicity (IC50 = 156 μM) similar to free 5CNU against HeLa cells upon visible light irradiation, [Ru(tpy)(CH3CN)3]2+ had a negligible impact on the viability of the cells under the same conditions, suggesting that only one 5CNU or one CH3CN is released due to a weak irradiation intensity and the toxicity stems from the released 5CNU rather than from the Ru fragments. Compared to the Ru complexes that can photo-inactivate cancer cells by the ROS mechanism, the ROS generation efficiency of 1 still needs improvement. Gasser and coworkers recently reported a new Ru complex, Ru-(DIP)2(bdt) (DIP = 4,7-diphenyl-1,10-phenanthroline and bdt = 1,2-benzenedithiolate) that has a 1O2 quantum yield of 0.81 in CH3CN and an IC50 value of 0.62 μM upon 420 nm irradiation (6.95 J cm−2) against HeLa cells.24
 | ||
| Fig. 10 Cytotoxicity of 1, 2 and py-Fc toward SKOV3 cells (upper) and A549 cells (lower) in the dark (black bar) and upon irradiation for 0.5 h (red bar). | ||
The non-luminescent feature of 1–3 excluded the use of the confocal fluorescence imaging techniques to determine their sub-cellular localization. Due to the multi-target character of ROS, the phototoxicity of 1 cannot definitely be attributed to DNA damage. Proteins, enzymes and lipid molecules can be possible targets as well.
As expected, 2 displayed similar cytotoxicity both in the dark and upon irradiation, in line with its inability for either ligand photo-dissociation or ROS generation. Interestingly, the photoactivation effect was not observed in the case of py-Fc even though it may generate ·OH. Similar behavior was also found in DNA experiments. The highly lipophilic character of py-Fc may, at least, partly account for the observations. On the one hand, this facilitates cellular uptake and improves the dark toxicity. On the other hand, this triggers aggregation and decreases the generation of ·OH.
The cellular uptake behaviors of 1 and 2 were assessed using inductively coupled plasma-mass spectrometry (ICP-MS). After incubation with A549 or SKOV3 cells for 4 h at 20 μM, 1 displayed an uptake value of 418 pmol/106 cells for A549 and 468 pmol/106 cells for SKOV3, respectively. Under the same conditions, no uptake of 2 was detected for both types of cells. The improved uptake of 1 with respect to 2 may be attributed to the incorporation of py-Fc in its structure, which enhances its lipophilicity as shown in Table 1. Due to the presence of the Fe element in cells and in their culture media, the ICP-MS measurement of py-Fc was not conducted.
Conclusions
A new Ru(II) arene complex with py-Fc as a monodentate ligand was designed and synthesized to achieve individual anticancer activity of each component simultaneously. 1 has a long-wavelength absorption band extending to 600 nm. Visible light irradiation led to monodentate ligand dissociation along with the generation of not only ·OH, but also 1O2. While the photoinduced intramolecular electron transfer from py-Fc to the Ru(II) arene makes a ligand exchange reaction possible, the photoinduced intermolecular electron transfer from py-Fc to O2 accounts for the ·OH generation ability of the complex. Moreover, the combination of the highly efficient ISC and heavy metal effect pertaining to a Ru(II) arene with the low-lying triplet excited state of py-Fc enables the complex with a new capability to generate 1O2. As a result, 1 can cleave plasmid DNA and inactivate SKOV3 and A549 cells upon light irradiation. All these results, and the underlying mechanisms, bring new thoughts for us to design new metallodrugs bearing dual activity from PACT and PDT.Experimental section
Materials
Bromoferrocene was purchased from the Tokyo Chemical Industry (TCI). 4-Pyridylboronic acid, Pd(PPh3)4, 2,2′-bipyridine, silver nitrate, anhydrous potassium carbonate, [{(η6-p-cymene)RuCl(μ-Cl)}2] and solvents of HPLC grade, such as methanol, dimethyl sulfoxide and acetonitrile, were purchased from Sigma-Aldrich.Syntheses
:CH3CH2COOC2H5 = 5:1. Yield = 80.6%.1H NMR (400 MHz, in CD3OD): δ = 4.05 (s, 5H), 4.49 (s, 2H), 4.89 (s, 2H), 7.52–7.53 (d, 2H, J = 7.5 Hz), 8.35–8.37 (d, 2H, J = 8.4 Hz). HR ESI-MS: calcd for (C15H14NFe)+, m/z = 264.0470; found, m/z = 264.0463.
:5:1) as an eluent. The compound was dissolved in H2O (2 mL) and precipitated by excess NH4PF6, and the orange solid was filtered, washed with water for three times and vacuum dried. Yield = 51.3%.1H NMR (400 MHz, in CD3OD): δ = 0.96–0.97, (d, 6H, J = 4.0 Hz), 1.93 (s, 3H), 2.51–2.58 (m, 1H), 3.98 (s, 5H), 4.59 (s, 2H), 4.85 (s, 2H), 6.12–6.13 (d, 2H, J = 4.0 Hz), 6.55–6.57 (d, 2H, J = 8.0 Hz), 7.44–7.45 (d, 2H, J = 4.0 Hz), 7.99–8.13 (t, 2H, J = 8.0 Hz), 8.11–8.13 (d, 2H, J = 8.0 Hz), 8.35–8.39 (t, 2H, J = 8.0 Hz), 8.52–8.54 (d, 2H, J = 8.0 Hz), 9.80–9.81 (d, 2H, J = 4.0 Hz). HR ESI-MS: calcd for (C35H35FeN3Ru)2+(M-2PF6)2+, m/z = 327.5607; found, m/z = 327.5603. Anal. calcd for C35H35F12FeN3P2Ru·H2O: C, 43.67; H, 3.87; N, 4.37. Found: C, 43.52; H, 3.90; N, 4.33.
Instruments and methods
1H NMR spectra were obtained on a Bruker DMX-400 MHz spectrophotometer, taking SiMe4 as a reference standard. High resolution electrospray ionization mass spectra (HR ESI-MS) were recorded on a Bruker APEX IV FT_MS. Elemental analysis was performed on an Elementar Vario EL instrument. UV-vis spectra were measured on a Shimadzu UV-1601PC spectrophotometer. Fluorescence emission spectra were taken on a Hitachi F-4600 fluorescence spectrophotometer.EPR spectra were obtained on a Bruker ESP-300E spectrometer at 9.8 GHz, X-band with 100 Hz field modulation, using TEMP and DMPO as spin trapping agents of 1O2 and ·OH, respectively. Samples were injected quantitatively into home-made quartz capillaries and illuminated in the cavity of the EPR spectrometer with a xenon lamp over 400 nm.
The electrochemical properties were measured on an EG&G Model283 potentiostat/galvanostat in a three-electrode cell with a glassy carbon working electrode, a Pt counter electrode, and a saturated calomel electrode (SCE) as reference. Cyclic voltammetry was operated at a scan-rate of 50 mV s−1 in N2-saturated, anhydrous CH3OH containing 0.1 M Bu4NPF6 as the supporting electrolyte.
Oil/water partition coefficient measurement
The n-octanol/water partition coefficients (logPO/W) were determined at room temperature following a reported method.26 Typically, solutions of each compound (20 μM) in 2 mL n-octanol and 2 mL PBS (pH = 7.4, 5 mM) were sonicated for 30 min. After separation by centrifugation, the concentrations of the compound in each phase were quantified by UV-vis spectroscopy. The results were the average of three independent measurements.
1O2 measurement
The reaction of 1O2 with DPBF was adopted to assess the 1O2 generation ability.22 A series of 2 mL of air-saturated CH3CN solutions of DPBF and the examined compound, of which the absorbance at 380 nm was adjusted to the same, were illuminated with 380 nm light (obtained from a Hitachi F-4600 fluorescence spectrophotometer). The consumption of DPBF was followed by recording the emission spectra of DPBF.·OH measurement
A 2 mL aqueous solution containing 1 mM terephthalic acid (TA), 2 mM NaOH and 100 μM 1 was irradiated at 470 nm (LED light source, 0.32 mW cm−2). The fluorescence emission spectra changes of the irradiated solution were monitored under the excitation wavelength of 315 nm.21DNA electrophoresis
DNA photo-cleavage abilities of the examined compounds were evaluated using a supercoiled pUC19 plasmid DNA as the target. A 50 μL PBS (pH = 7.4, 5 mM) solution of DNA (40 μg mL−1) and the examined compound (different concentrations) was irradiated under an Oriel 91192 solar simulator with a long-pass glass filter of 400 nm. After irradiation, 10 μL gel loading buffer was added. A 10 μL sample was loaded on agarose gel electrophoresis (in Tris–acetic acid–EDTA buffer, pH 8.0) at 80 V for 1 hour. The gel was then stained with ethidium bromide (1 mg L−1 in H2O) for 0.5 h, washed with water twice and then analyzed using a Gel Doc XR system (Bio-Rad).Cytotoxicity assay
The MTT assay method was applied to analyze cell viability. SKOV3 or A549 cells were plated in 2 × 105 per well in a Nunc 96 well plate and incubated for 24 h in 150 μL low glucose DMEM medium with FBS at 37 °C under 5% CO2 atmosphere. Then, the cells were exposed to new DMEM medium without FBS containing different concentrations of the examined compounds and incubated for 4 h at 37 °C. Next, the cells were activated with light 470 nm (from a LED light source, 0.32 mW cm−2) at 25 °C for 30 min. After another 20 h of incubation in the dark at 37 °C, MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) solution in DMEM without FBS was added and the cells were maintained at 37 °C for 4 h. After removal of the medium, a mixed solution of CH3OH/DMSO (1:1) was added and the absorbance at 570 nm was read on a Thermo MK3 Multiscan microplate reader. The cell viability data were normalized to 100% viable (untreated) cells and were the average of at least three independent measurements at each dose.
Cellular uptake
A549 and SKOV3 cells were planted at a concentration of 1 × 105 cells per mL in 25 cm2 cell culture flasks. After 12 h incubation, the medium was removed and the cells were treated with 20 μM complexes in fresh medium without FBS for 4 h. The medium was removed again and the cells were washed with PBS three times and harvested by trypsinization. The cells was counted by a hemocytometer and then collected by centrifugation at 5000 rpm for 5 min. Pellets were digested and analysed on a NexION 300X inductively coupled plasma mass spectrometer (ICP-MS).Acknowledgements
This work was financially supported by the Ministry of Science and Technology (2013CB933801) and NSFC (21390400, 21172228, 21273259, 21571181, 21301182).Notes and references
-
(a) N. P. E. Barry and P. J. Sadler, Chem. Commun., 2013, 49, 5106–5131 RSC
; (b) G. Sava, A. Bergamo and P. J. Dyson, Dalton Trans., 2011, 40, 9069–9075 RSC
-
(a) A. Gandioso, E. Shaili, A. Massaguer, G. Artigas, A. González-Cantó, J. A. Woods, P. J. Sadler and V. Marchán, Chem. Commun., 2015, 51, 9169–9172 RSC
- F. S. Mackay, J. A. Woods, P. Heringova, J. Kašpárková, A. M. Pizarro, S. A. Moggach, S. Parsons, V. Brabec and P. J. Sadler, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 20743–20748 CrossRef CAS PubMed
-
(a) Q. Zhou, Y. Zheng, T. Wang, Y. Chen, K. Li, Y. Zhang, C. Li, Y. Houa and X. Wang, Chem. Commun., 2015, 51, 10684–10686 RSC
-
(a) W.-Q. Cao, W.-J. Zheng and T.-F. Chen, Sci. Rep., 2015, 5, 9157 CrossRef CAS PubMed
-
(a) J. D. Knoll, B. A. Albani and C. Turro, Acc. Chem. Res., 2015, 48, 2280–2287 CrossRef CAS PubMed
-
(a) Y. Zheng, Q. Zhou, Y. Zhang, C. Li, Y. Hou and X. Wang, Dalton Trans., 2016, 45, 2897–2905 RSC
-
(a) M. R. Detty, S. L. Gibson and S. J. Wagner, J. Med. Chem., 2004, 47, 3897–3915 CrossRef CAS PubMed
-
(a) G. S. Smith and B. Therrien, Dalton Trans., 2011, 40, 10793–10800 RSC
- S. Betanzos-Lara, L. Salassa, A. Habtemariam and P. J. Sadler, Chem. Commun., 2009, 6622–6624 RSC
- Q. Zhou, W. Lei, Y. Hou, Y. Chen, C. Li, B. Zhang and X. Wang, Dalton Trans., 2013, 42, 2786–2791 RSC
- M. F. R. Fouda, M. M. Abd-Elzaher, R. A. Abdelsamaia and A. A. Labib, Appl. Organomet. Chem., 2007, 21, 613–625 CrossRef CAS
-
(a) C. S. Allardyce, A. Dorcier, C. Scolaro and P. J. Dyson, Appl. Organomet. Chem., 2005, 19, 1–10 CrossRef CAS
-
(a) W. E. Butler, P. N. Kelly, A. G. Harry, R. Tiedt, B. White, R. Devery and P. T. M. Kenny, Appl. Organomet. Chem., 2013, 27, 361–365 CrossRef CAS
- A. Leonidova, P. Anstaett, V. Pierroz, C. Mari, B. Spingler, S. Ferrari and G. Gasser, Inorg. Chem., 2015, 54, 9740–9748 CrossRef CAS PubMed
-
(a) J. Wang, Y. Hou, W. Lei, Q. Zhou, C. Li, B. Zhang and X. Wang, ChemPhysChem, 2012, 13, 2739–2747 CrossRef CAS PubMed
- G. Tabbı, C. Cassino, G. Cavigiolio, D. Colangelo, A. Ghiglia, I. Viano and D. Osella, J. Med. Chem., 2002, 45, 5786–5796 CrossRef
- K. Reszka, F. S. Cruz and R. Docampo, Chem.–Biol. Interact., 1986, 58, 161–172 CrossRef CAS PubMed
-
(a) C. Hadjur, A. Jeunet and P. Jardon, J. Photochem. Photobiol., B, 1994, 26, 67–74 CrossRef CAS
- E. J. Lee and M. S. Wrighton, J. Am. Chem. Soc., 1991, 113, 8562–8564 CrossRef CAS
- C. Fang, H. Jia, S. Chang, Q. F. Ruan, P. Wang, T. Chen and J. F. Wang, Energy Environ. Sci., 2014, 7, 3431–3438 CAS
- R. H. Young, K. Wehrly and R. L. Martin, J. Am. Chem. Soc., 1971, 93, 5774–5779 CrossRef CAS
- A. A. Abdel-Shafi, P. D. Beer, R. J. Mortimer and F. Wilkinson, J. Phys. Chem. A, 2000, 104, 192–202 CrossRef CAS
- A. Frei, R. Rubbiani, S. Tubafard, O. Blacque, P. Anstaett, A. Felgenträger, T. Maisch, L. Spiccia and G. Gasser, J. Med. Chem., 2014, 57, 7280–7292 CrossRef CAS PubMed
- C. Imrie, C. Loubser, P. Engelbrecht and C. W. McCleland, J. Chem. Soc., Perkin Trans. 1, 1999, 2513–2523 RSC
- M. Kepczynski, R. P. Pandian, K. M. Smith and B. Ehrenberg, Photochem. Photobiol., 2002, 76, 127–134 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: absorption spectrum protonated py-Fc, CV of 1, 2, and py-Fc, EPR signals of py-Fc, fluorescence spectra of TA and 1, agarose gel electrophoresis of 2 and py-Fc. See DOI: 10.1039/c6ra05182k |
| This journal is © The Royal Society of Chemistry 2016 |