
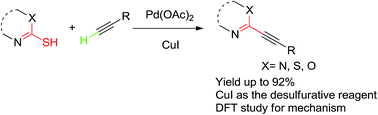
CuI assisted desulfurative Sonogashira reaction of mercapto N-heterocyclic derivatives with alkynes†
Abstract
A palladium catalyzed Sonogashira reaction of mercapto N-heterocyclic derivatives with terminal alkynes by using CuI as the desulfurative reagent was described in this report. It provided an effective strategy for obtaining sp–sp2 cross-coupling products in good yields. The reaction mechanism was also investigated by density functional theory (DFT) calculation.
 Please wait while we load your content...
Please wait while we load your content...

