
Dioxotungsten(vi) complexes with isoniazid-related hydrazones as (pre)catalysts for olefin epoxidation: solvent and ligand substituent effects†
 *bcd
*bcd
Abstract
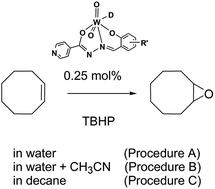
The mononuclear dioxotungsten(VI) complexes [WO2(L3OMe)(D)] (1a and 1b), [WO2(L4OMe)(D)] (2a and 2b) and [WO2(LH)(D)] (3a and 3b) (D = EtOH (1a–3a) or MeOH (1b–3b); L3OMe = 3-methoxy-2-oxybenzaldehyde isonicotinoyl hydrazonato, L4OMe = 4-methoxy-2-oxybenzaldehyde isonicotinoyl hydrazonato, LH = 2-oxybenzaldehyde isonicotinoyl hydrazonato) were synthesized by the reaction of [WO2(acac)2]·0.5C6H5Me with the respective isoniazid-related hydrazone. The compounds were characterized by microanalysis, FT-IR and NMR spectroscopy, thermogravimetric analysis, and powder X-ray diffraction method. The crystal and molecular structures of 1a, 1b, 3a and [WO2(acac)2]·0.5C6H5Me were determined by single crystal X-ray diffraction. The structures of 1a, 1b, 3a are mononuclear and form hydrogen bonded centrosymmetric dimers. In all three complexes, the dimers are also held together by π⋯π interactions between aromatic rings. The catalytic performances (activity and selectivity) of 1a–3a and 1b–3b towards alkene epoxidation by tert-butyl hydroperoxide (TBHP) were investigated under different conditions.
 Please wait while we load your content...
Please wait while we load your content...

