
Enhanced UV detection by transparent graphene oxide/ZnO composite thin films
Abstract
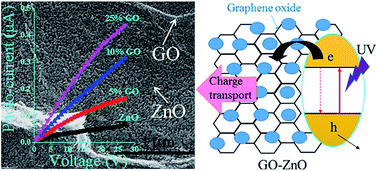
All solution processed transparent thin films of graphene oxide (GO) and zinc oxide (ZnO) in different compositions prepared by a simple two-step chemical synthesis method have been studied for their UV detection properties. The preparation of GO through oxidation of graphite flakes is followed by sol–gel spin coating deposition of the GO–ZnO composite films on glass substrates. The surface morphology, microstructure and composition of the samples have been studied to confirm the formation of composite thin films comprising wurtzite-ZnO nanocrystallites and GO flakes. Optical studies demonstrate that both the transparency and optical band gap of the samples as estimated from wavelength dependent transmittance curves decrease with the increase of GO content in the films, while the charge carrier concentration increases by 5 fold. The in-plane current–voltage (I–V) measurements with two silver electrodes on the GO–ZnO film show a significant enhancement of the photosensitivity in comparison to ZnO films when they are exposed to UV light of different intensities. The response time (t90-response) is nearly three times smaller for GO–ZnO composite films as compared to that of pure ZnO. This improvement is attributed to the defect state modulation and carrier density improvement of the thin films with incorporation of GO, which is encouraging to propel optical, electrical and hence optoelectronics applicability of ZnO composite based transparent devices.
 Please wait while we load your content...
Please wait while we load your content...

