DOI:
10.1039/C6RA05000J
(Paper)
RSC Adv., 2016,
6, 59327-59332
Facile synthesis of functional poly(vinylene sulfide)s containing donor–acceptor chromophores by a double click reaction†
Received
25th February 2016
, Accepted 10th June 2016
First published on 14th June 2016
Abstract
The new electronically active poly(vinylene sulfide)s containing dialkylaniline-substituted electron-rich alkynes in the side chains were designed and synthesized by the ‘thiol-click’ polymerization. Subsequently, the donor–acceptor chromophores were introduced into the side chains of the polymer by the [2 + 2] click reaction as an efficient postfunctionalization method. The polymers showed good solubility in common organic solvents and high thermal stability. The photophysical and electrochemical properties, as well as the click reactions, were characterized by means of UV-vis absorption spectroscopy, cyclic voltammetry, etc. After the introduction of donor–acceptor chromophores, the polymer showed a strong charge-transfer (CT) band in the visible absorption region, potent redox activities and a narrow band gap compared with the precursors. In addition, the third-order nonlinear properties, including the nonlinear absorption and the nonlinear susceptibilities, were investigated by using Z-scan techniques. A typical saturable absorption behavior was observed for the third order nonlinear absorption, with the nonlinear absorption coefficient (β) values of the polymer being −9.0 × 10−12 m W−1.
Introduction
Conjugated polymers offer great economic advantages as low-cost electronic conducting materials for the production of large-area flexible optoelectronic devices such as light-emitting diodes, organic photovoltaic cells (OPVs), and field-effect transistors.1–14 In conjugated polymers, the sulfur-containing π-conjugated polymers are of particular scientific interest as they may create new functionalities that are difficult, if not impossible, to access using polymers with pure carbon-based skeletons.15–20 Heretofore among all the sulfur-containing π-conjugated polymers, polythiophene based organic OPVs and poly(phenylene sulfide) have been extensively studied because of their excellent optoelectronic properties.15–20 However, the new types of electronically active poly(vinylene sulfide)s, which incorporate a carbon–carbon double bond between the aromatic ring and the sulfur atom in the polymer repeat unit, have been rarely reported.21,22
In the structures of poly(vinylene sulfide)s, the delocalization of π-electrons through the sulfur bridge may endow the polymers with an array of remarkable characteristics, such as light refractivity, nonlinear optical property and so on.21,22 However, the relatively low charge delocalization and large band gap still severely limit the improvement of the optoelectronic properties of poly(vinylene sulfide)s. Fortunately, the formal [2 + 2] click reactions between electron alkynes and electron-deficient alkenes, such as tetracyanoethylene (TCNE) or 7,7,8,8-tetracyanoquinodimethane (TCNQ), have been developed as a convenient and robust method for preparing narrow band gap π-conjugated polymers containing donor–acceptor chromophores.23–29 The reactions are with significant advantages that the products feature strong charge-transfer (CT) interactions in the visible absorption region, potent redox activities, and are useful for optimization of the electronic states, thereby leading to the enhanced performance of the optoelectronic devices, such as photovoltaic cells.30,31
Herein, the new electronically active poly(vinylene sulfide)s were synthesized by the thiol-yne click reaction.32–35 Moreover, the novel [2 + 2] click reaction was selected to postfunctionalize the poly(vinylene sulfide)s. It was envisioned that the [2 + 2] click reaction may efficiently modulate the HOMO–LUMO energy band gap and improve the optoelectronic properties of poly(vinylene sulfide)s.
Results and discussion
Synthesis
The 4,4′-thiobisbenzenethiol M2 and 4-((2,5-diethynylphenyl)ethynyl)-N,N-dihexadecylaniline M1 were designed as monomers for polymerization. As shown in Scheme 1, M1 has two kinds of activated alkynes. One is the terminal alkyne, and it can be reacted with thiol to form the sulfide-containing acetylenic polymers. Another is the side alkyne, and it can be reacted with the special click reagents such as TCNE, TCNQ, etc. ([2 + 2] click reaction).23–29 Thus poly(vinylene sulfide)s containing internal electron-rich alkynes in the side chains was prepared by ‘thiol-click’ polymerization and postfunctionalized by the acceptor TCNE.
 |
| | Scheme 1 Synthesis of poly(vinylene sulfide)s P1 and the postfunctionalization by TCNE addition. (a) Rh(PPh3)3Cl, DCE, 60 °C, 24 h, Ar; (b) TCNE, DCM, rt, 2 h. | |
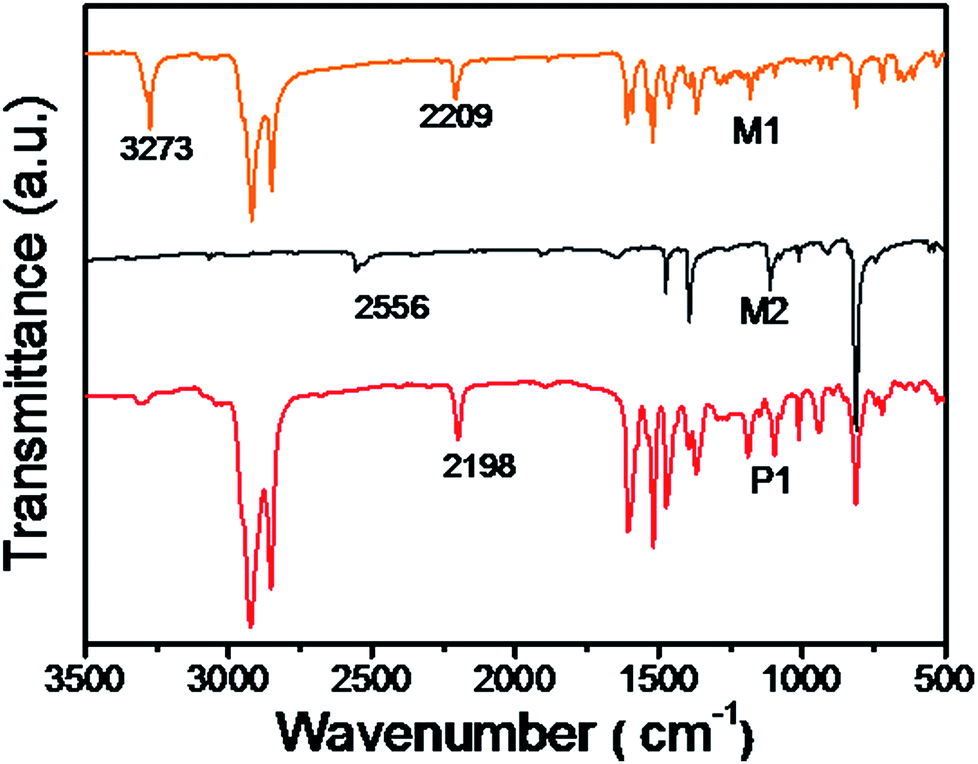
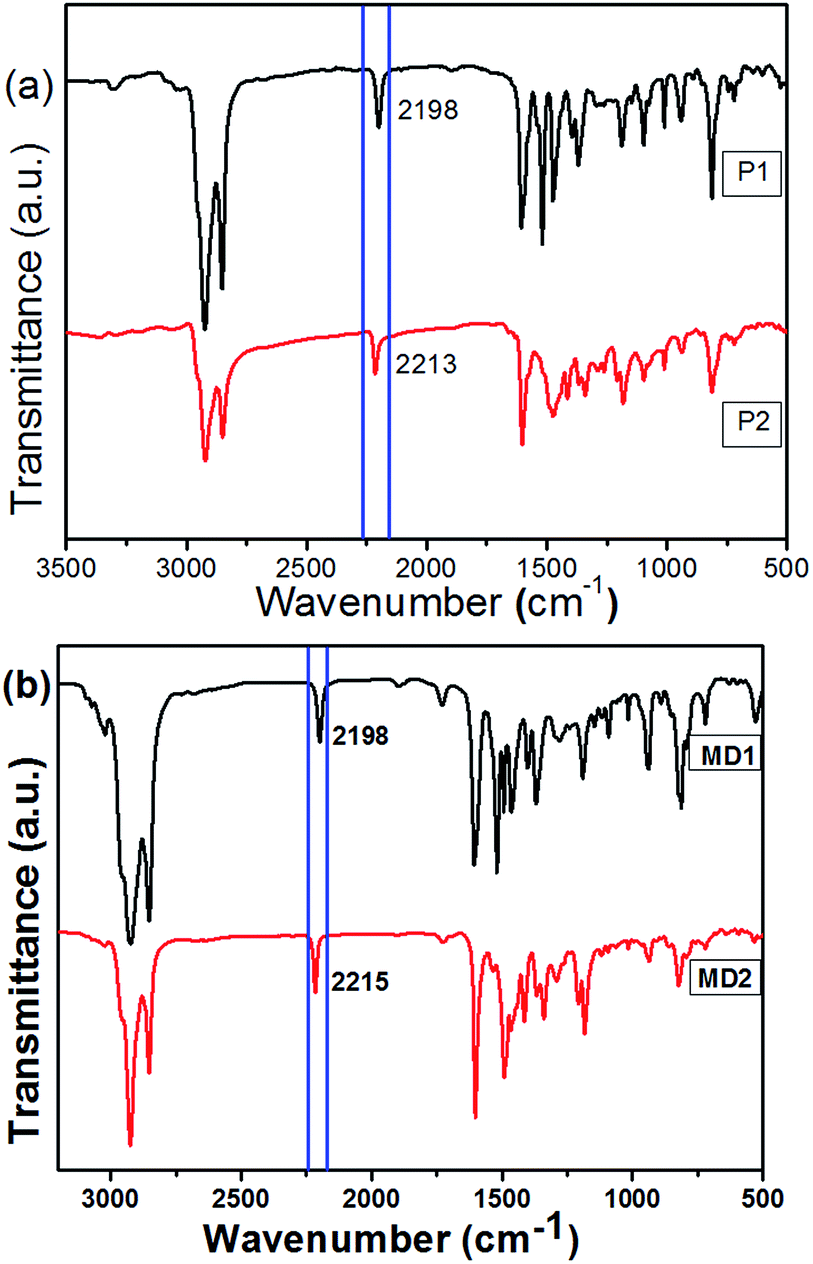
The chemical structures of P1 and P2 were substantiated by the combination of 1H-NMR and IR. In the 1H-NMR, peaks at 3.39 and 3.13 ppm ascribed to the terminal alkynes of M1 completely disappeared after the polymerization. As compared to MD1 and MD2, the spectra of P1 and P2 were obviously broadened. It's worth noting that the monomers M1 are asymmetric, so the obtained polymers should contain different regioregularity. It is clear that the polymers should contain all head-to-head, head-to-tail, and tail-to-tail conformations. Up to date, there is no report to prove the chemical structures.36,37 In addition, as shown in Fig. 1, the stretching vibration bands of ![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–H at 3273 cm−1 and CC and –SH at 2556 cm−1 in the spectra of monomers became much weaker after polymerization, indicating the generation of thiol-yne click addition. Moreover, as shown in Fig. 2(a), the alkyne vibration peak at 2198 cm−1 for P1 was replaced by strong cyano peaks at 2213 cm−1 for P2 supported the TCNE addition.
C–H at 3273 cm−1 and CC and –SH at 2556 cm−1 in the spectra of monomers became much weaker after polymerization, indicating the generation of thiol-yne click addition. Moreover, as shown in Fig. 2(a), the alkyne vibration peak at 2198 cm−1 for P1 was replaced by strong cyano peaks at 2213 cm−1 for P2 supported the TCNE addition.
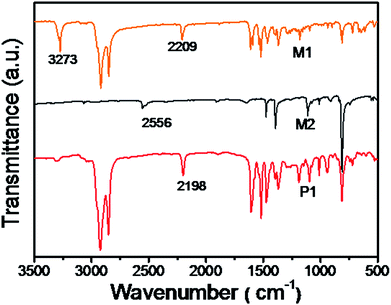 |
| | Fig. 1 FT-IR spectra of monomers M1, M2 and polymer P1. | |
 |
| | Fig. 2 FT-IR spectra of (a) polymers P1 and P2; (b) model compounds MD1 and MD2. | |
The molecular weights of polymers were shown in Table 1. The molecular weight (Mn) and the polydispersity (Mw/Mn) of P1 and P2 determined by GPC relative to standard polystyrenes were 7200 and 3.40, 8000 and 3.70, respectively. In addition, both P1 and P2 displayed good solubilities in the common organic solvents, such as DCM, DCE and THF. It can be attributed to the introduction of the long alkyl chains of hexadecyl groups.
Table 1 Summary of molecular weight and thermal properties
| Polymer |
Mna |
Mw |
Mw/Mn |
Tgb/°C |
T5%c/°C |
T10%c/°C |
| Determined by GPC (THF eluent, calibrated by polystyrene standards). Glass transition temperature determined by the second heating scan of DSC measurements. Temperature at which 5% and 10% weight loss occurred upon heating. |
| P1 |
7200 |
24![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 480 480 |
3.4 |
— |
304 |
324 |
| P2 |
8000 |
29600 |
3.7 |
— |
245 |
285 |
Model compounds were usually designed and prepared to prove the chemical structures and investigate the photoelectric properties.38–42 Therefore correspond model compounds of the polymers were synthesized, and the molecular structures were shown in Scheme 2. Both the model compounds were characterized by 1H-NMR, IR, MS spectra and elemental analysis. The 1H-NMR of the compounds MD1 and MD2 showed multiplet peaks in the aromatic region. The new peak appeared at 2.67 ppm ascribed to the thiol proton supported the combination between monomers M1 and M2. In addition, the peaks in the aromatic region became more complex after TCNE addition. Moreover, as shown in Fig. 2(b), the alkyne vibration peak at 2198 cm−1 for MD1 was replaced by strong cyano peaks at 2215 cm−1 for MD2 also supported the TCNE addition. These results, in combination with other characterization data, strongly supported the clean formation of model compounds and polymers.
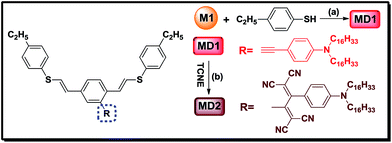 |
| | Scheme 2 Syntheses model compounds MD1 and MD2, (a) Rh(PPh3)3Cl, DCE, 60 °C, 15 h, Ar; (b) TCNE, DCM, rt, 2 h. | |
Thermal properties
Thermal stability of the polymers was investigated by thermogravimetric analysis (TGA) at the scanning rate of 10 °C min−1 under flowing nitrogen. In contrast to the previous report that the [2 + 2] click reaction sometimes enhances the thermal stability of polymers and materials,42–44 the 5% decomposition temperatures (T5%) of the polymer P1 was changed from 304 °C to 245 °C after TCNE addition (in Fig. 3 and Table 1). The result may be derived from reduced individual bond energy by introducing strong acceptor cyano groups. In addition, the glass transition temperatures of the polymers were estimated by the DSC measurements at the scanning rate of 10 °C min−1 under flowing nitrogen. Unexpectedly, both the polymers P1 and P2 did not show any well-defined transition in the range from 20 °C to 250 °C (in Fig. 3: inset).
 |
| | Fig. 3 Thermogravimetric analysis of P1 and P2; inset: DSC curves of polymers P1 and P2. | |
UV-vis spectroscopy
To estimate the progress of the reactions as well as the presence of side reactions, the titration experiments of the model compound MD1 with TCNE were performed and the UV-vis spectral change was monitored. The UV-vis spectral change was shown in Fig. 4. When quantitative TCNE was added to the dichlorobenzene solution of MD1, the addition reaction rapidly proceeded at rt with the solution color changing. Moreover, as more TCNE was added, the original peaks centered at 342 nm decreased, whereas a new peak appeared and increased at 492 nm. The intensity of the new peak linearly increased and the presence of an isosbestic point at 378 nm indicated the absence of any undesired side reactions.
 |
| | Fig. 4 Normalized UV-vis spectral change of MD1 in 1,2-dichlorobenzene during the titration experiment with a TCNE solution in DCM at rt. Inset: plots of TCNE addition amount versus absorbance increase at 492 nm. | |
The characteristic UV-vis absorption spectra and the solution color of the polymers and model compounds in DCM were shown in Fig. 5. The spectra of polymers were almost the same as that of the corresponding model compounds, and the studies indirectly proved that polymers have the similar chemical structures with the model compounds. After the TCNE addition, as shown in Fig. 5 (inset), the solution colors of the P1 and MD1 changed from light yellow to reddish. In addition, the end absorption (λend) of P1 and MD1 bathochromically shifted to more than 800 nm. Both the click-type reaction products P2 and MD2 displayed two distinct bands in the absorption spectra. One band at shorter wavelengths region (300–400 nm) is due to localized π–π* transitions (LT), and the other band at longer wavelengths (400–800 nm) is attributed to intramolecular charge transfer (ICT) band between electron-rich donors and electron-deficient acceptors.23–29 However, the LT and ICT bands of P2 and MD2 were overlapped into a broad band covering the whole visible region from 400 to 800 nm, indicating that the click moieties were the strong electron-withdrawing groups.
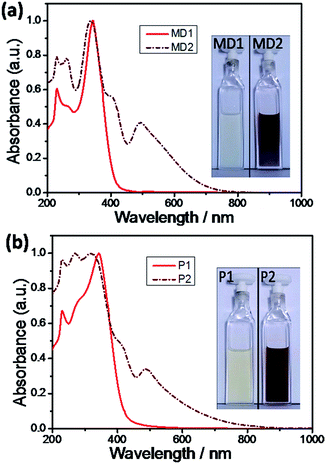 |
| | Fig. 5 Normalized UV-vis spectra of (a) MD1 and MD2, (b) P1 and P2 in DCM. Inset: pictures of P1, P2, MD1 and MD2 solutions in DCM. | |
Electrochemistry
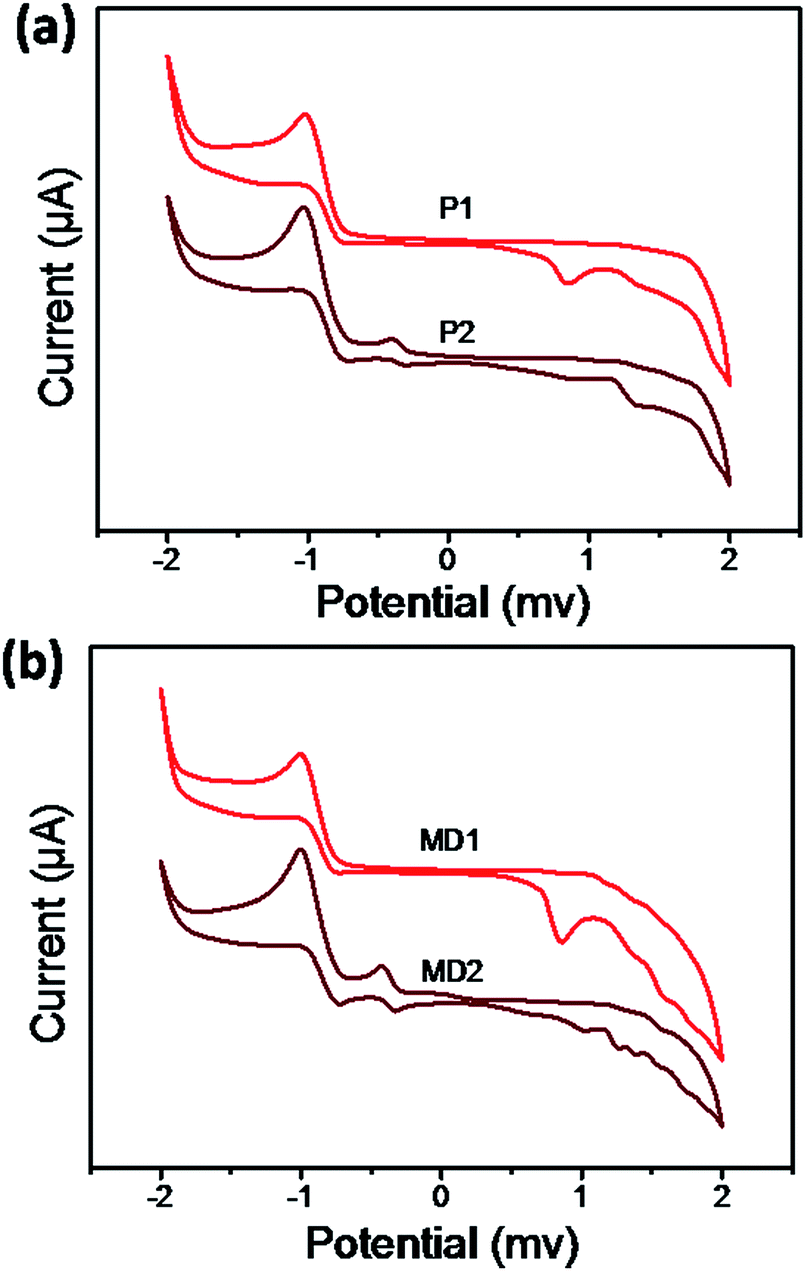
One of the most important features of donor–acceptor chromophores was the oxidation/reduction activities.45 In order to reveal the electrochemical properties and estimate their energy levels, electrochemical measurements were performed. The cyclic voltammograms were shown in Fig. 6 and onset oxidation/reduction potentials, band gaps, LUMO and HOMO levels were listed in Table 2. Both the polymer P1 and compound MD1 showed the only irreversible oxidation peaks ascribed to the dialkylaniline moieties in the side chains before the addition of TCNE. However, the products P2 and MD2 showed anodically shift (Eoxon) at 1.13 and 1.10 V, as well as the first reduction potentials (Eredon) at −0.27 and −0.31 V, respectively after TCNE addition. In addition, the calculated electrochemical band gaps of the click-type reaction products P2 and MD2 were 1.40 eV and 1.41 eV, which were in fair agreement with the corresponding optical band gaps determined from the λend values. Moreover, the band gaps of P2 and MD2 were decreases compared with P1 and MD1 due to the introduction of the acceptor cyano groups by means of click-type reactions. Meanwhile, both the HOMO and LUMO levels decreased as acceptor TCNE was added. In particular, the extent of the decrease in the LUMO levels was more significant compared with the HOMO levels. It is consistent with a fact that the alkyne groups feature an electron-accepting ability and thereby affects the LUMO levels.46 The HOMO energy levels of P1, and P2 were −5.29 and −5.72 eV, respectively, implying that they varied with respect to the modulated ICT strengths resulting from the presence of electron donors with various electron donating abilities.47 The LUMO energy levels of P2 and MD2 were all located within a reasonable range (around −3.0 eV, Table 2) and were significantly lower than that of PC61BM (ca. 4.1 eV); thus, the efficient charge transfer/dissociation to PC61BM would be prevented. Moreover, low HOMO levels and narrow energy gaps suggest that all compounds are promising strong electronic transition.48
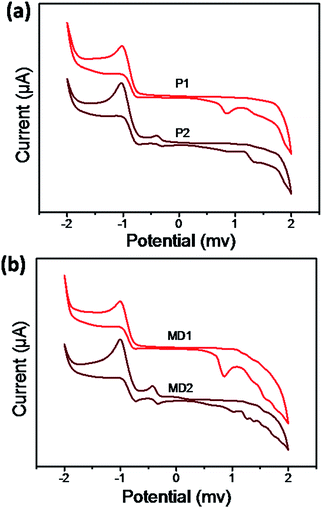 |
| | Fig. 6 Cyclic voltammograms of (a) P1, P2 and (b) MD1, MD2 in DCM. | |
Table 2 Summary of optical and electrochemical data
| Materials |
P1 |
P2 |
MD1 |
MD2 |
| Measured in DCM at 20 °C. Determined by the λend values. Measured in DCM with 0.1 M Bu4NPF6 at 20 °C. Potentials vs. Fc/Fc+. Determined by the Eonsetox or Eonsetred values based on the assumption of Fc/Fc+ = −4.80 ev. Calculated from the HOMO and optical band gap. |
| λend (nm)a |
580 |
890 |
528 |
784 |
| Eg (ev)b |
2.14 |
1.39 |
2.35 |
1.58 |
| Eoxon (V)c |
0.70 |
1.13 |
0.71 |
1.10 |
| Eredon (V)c |
— |
−0.27 |
— |
−0.31 |
| Eg (ev)c |
— |
1.40 |
— |
1.41 |
| HOMO (ev)d |
−5.29 |
−5.72 |
−5.30 |
−5.69 |
| LUMO (ev) |
−3.15e |
−4.32d |
−2.95e |
−4.28d |
| β (10−12 m W−1) |
— |
−9.0 |
— |
−4.8 |
| χ(3) (10−13 esu) |
— |
1.37 |
— |
0.73 |
Nonlinear optics
In investigations of the third-order nonlinear optical properties, the third-order susceptibility χ(3) of all products were measured by means of Z-scan technique. Although the precursors P1 and MD1 did not show any obvious signal in the open Z-scan curves, the click-reaction products P2 and MD2 exhibited the similar classic saturable absorption (SA) behaviors, as shown in Fig. 7. The nonlinear absorption coefficients (β) values of P2 and MD2 were −9.0 × 10−12 m W−1 and −4.8 × 10−12 m W−1 (in Table 2). By contrast, no nonlinear refraction was displayed in the closed Z-scan measurement of all products. The calculated third-order susceptibility χ(3) of P2 and MD2 were 1.37 × 10−13 esu and 0.73 × 10−13 esu. The generate of the NLO properties could be attributed to the existence of donor–acceptor chromophores separated by a π-conjugated systems in the P2 and MD2 structures, which could be efficient guidance for NLO molecular designs.
 |
| | Fig. 7 The Z-scan experimental and theoretical dates of open apertures of P2 and MD2. | |
We got the similar nonlinear optical properties for other derivatives such as pyrenes, triphenylenes, fullerenes, porphyrins, and so on.25,27,28,49,50 For all the compounds, the compounds with TCNE click moieties would show the SA behaviours.25,27,28,49,50 The conjugated polymer showed better nonlinear optical properties than those of small molecular, because of the longer conjugated structures.25,27 As we known, the fullerenes and porphyrins are the excellent nonlinear optical materials. Furthermore, the polymer P2 showed the same order of magnitude as those of fullerenes and porphyrins in the ref. 28, 49 and 50. Another polymer series with double click moieties were studied by us,51 and the flexible chains (–(CH2)6–) were designed in the main chain, the solubility should be better, but the conjugated structures were destroyed. In this paper, the different click chemistry method was selected (named ‘thiol-click’), and the whole conjugated structures for main chains were designed. We hope better NLO properties because of the longer conjugated structures, but the similar results were obtained here. It was suggested that the moieties from [2 + 2] click reagents affected greater than those of conjugated structures.
Conclusions
In summary, the combination of the conventional thiol-yne click reaction and alkyne-acceptor [2 + 2] click reaction was established to prepare the new electronically active sulfur-containing p-conjugated poly(vinylene sulfide)s. Investigations of the photophysical and electrochemical properties of the polymers by using UV-vis spectroscopy and cyclic voltammetry showed a strong intramolecular CT interactions and a relatively narrow band gap due to the formation of the donor–acceptor chromophores by the [2 + 2] click reaction. Most importantly, Z-scan results demonstrated that the poly(vinylene sulfide)s containing strong donor–acceptor chromophores may endow the polymer with the third-order nonlinear optical properties due to the adjustment of electron density distribution within the chromophores. Therefore the [2 + 2] click reaction provides a very effective method to improve the optoelectronic properties of poly(vinylene sulfide)s.
Acknowledgements
This work was supported by the National Key Basic Research Program of China (2014CB931804), the National Natural Science Foundation of China (Grant No. 51173017, 51373024, 51473020, 61370048), the Beijing Higher Education Young Elite Teacher Project (Grant No. YETP0356), Fundamental Research Funds for the Central Universities (Grant No. FRF-TP-14-001A2). Dong Wang and Qingsen Guo are the Equal Contribution.
Notes and references
- C. Sekine, Y. Tsubata, T. Yamada, M. Kitano and S. Doi, Sci. Technol. Adv. Mater., 2014, 15, 34203–34217 CrossRef PubMed.
- A. C. Grimsdale, K. L. Chan, R. E. Martin, P. G. Jokisz and A. B. Holmes, Chem. Rev., 2009, 109, 897–1091 CrossRef CAS PubMed.
- S. C. Lo and P. L. Burn, Chem. Rev., 2007, 107, 1097–1116 CrossRef CAS PubMed.
- J. S. Wu, S. W. Cheng, Y. L. Cheng and C. S. Hsu, Chem. Soc. Rev., 2015, 44, 1113–1154 RSC.
- H. J. Jhuo, P. N. Yeh, S. H. Liao, Y. L. Li, Y. S. Cheng and S. A. Chen, J. Chin. Chem. Soc., 2014, 61, 115–126 CrossRef CAS.
- L. M. Chen, Z. R. Hong, G. Li and Y. Yang, Adv. Mater., 2009, 21, 1434–1449 CrossRef CAS.
- S. Allard, M. Forster, B. Souharce, H. Thiem and U. Scherf, Angew. Chem., Int. Ed., 2008, 47, 4070–4098 CrossRef CAS PubMed.
- A. R. Murphy and J. M. J. Frechet, Chem. Rev., 2007, 107, 1066–1096 CrossRef CAS PubMed.
- W. L. Ma, C. Y. Yang, X. Gong, K. Lee and A. J. Heeger, Adv. Funct. Mater., 2005, 15, 1617–1622 CrossRef CAS.
- G. Li, V. Shrotriya, J. S. Huang, Y. Yao, T. Moriarty, K. Emery and Y. Yang, Nat. Mater., 2005, 4, 864–868 CrossRef CAS.
- Y. Kim, S. Cook, S. M. Tuladhar, S. A. Choulis, J. Nelson, J. R. Durrant, D. D. C. Bradley, M. Giles, I. McCulloch and C. S. Ha, Nat. Mater., 2006, 5, 197–203 CrossRef CAS.
- S. H. Park, A. Roy, S. Beaupre, S. Cho, N. Coates, J. S. Moon, D. Moses, M. Leclerc, K. Lee and A. J. Heeger, Nat. Photonics, 2009, 3, 297–302 CrossRef CAS.
- W. Huang, H. Y. Zhu, Y. L. Huang, J. W. Yang and W. Z. Wang, RSC Adv., 2016, 6, 40343–40353 RSC.
- A. Brzeczek, K. Piwowar, W. Domagala, M. M. Mikolajczyk, K. Walczak and P. Wagner, RSC Adv., 2016, 6, 36500–36509 RSC.
- A. P. Gies, J. F. Geibel and D. M. Hercules, Macromolecules, 2010, 43, 943–951 CrossRef CAS.
- P. Huo and P. Cebe, J. Polym. Sci., Part B: Polym. Phys., 1992, 30, 239–250 CrossRef CAS.
- L. H. Perng, Polym. Degrad. Stab., 2000, 69, 323–332 CrossRef CAS.
- P. Piaggio, D. Giribone, G. F. Musso, C. Cuniberti, G. Dellepiane and A. Borghesi, Synth. Met., 1991, 41, 1359–1364 CrossRef CAS.
- C. Y. Chang, S. P. Ju, J. W. Chang, S. C. Huang and H. W. Yang, RSC Adv., 2014, 4, 26074–26080 RSC.
- M. L. Zhang, H. X. Wang, Z. H. Li and B. W. Cheng, RSC Adv., 2015, 5, 13840–13849 RSC.
- B. C. Yao, J. Mei, J. Li, J. Wang, H. Q. Wu, J. Z. Sun, A. J. Qin and B. Z. Tang, Macromolecules, 2014, 47, 1325–1333 CrossRef CAS.
- B. C. Yao, T. Hu, H. K. Zhang, J. Li, J. Z. Sun, A. J. Qin and B. Z. Tang, Macromolecules, 2015, 48, 7782–7791 CrossRef CAS.
- M. Kivala and F. Diederich, Acc. Chem. Res., 2009, 42, 235–248 CrossRef CAS PubMed.
- S. Kato and F. Diederich, Chem. Commun., 2010, 46, 1994–2006 RSC.
- Y. S. Mi, P. X. Liang, Z. K. Jin, D. Wang and Z. Yang, ChemPhysChem, 2013, 14, 4102–4108 CrossRef CAS PubMed.
- D. Wang, W. S. Zhang, Y. Xing, H. Gao, X. K. Wang, Y. Z. Zhao and H. Yang, Tetrahedron, 2013, 69, 895–901 CrossRef CAS.
- Z. K. Jin, D. Wang, X. K. Wang, P. X. Liang, Y. S. Mi and H. Yang, Tetrahedron Lett., 2013, 54, 4859–4864 CrossRef CAS.
- W. S. Zhang, D. Wang, H. Cao and H. Yang, Tetrahedron, 2014, 70, 573–577 CrossRef CAS.
- P. X. Liang, Z. C. Du, D. Wang, Z. Yang, H. Y. Sheng, S. Q. Liang, H. Cao, W. L. He and H. Yang, ChemPhysChem, 2014, 15, 3523–3529 CrossRef CAS PubMed.
- T. Otsubo, Y. Aso and K. Takimiya, J. Mater. Chem., 2002, 12, 2565–2575 RSC.
- C. Tang, F. Liu, Y. J. Xia, J. Lin, L. H. Xie, G. Y. Zhong, Q. L. Fan and W. Huang, Org. Electron., 2006, 7, 155–162 CrossRef CAS.
- A. B. Lowe, Polym. Chem., 2014, 5, 4820–4870 RSC.
- M. N. Ganivada, P. Kumar and R. Shunmugam, RSC Adv., 2015, 5, 50001–50004 RSC.
- O. Z. Durham, H. R. Norton and D. A. Shipp, RSC Adv., 2015, 5, 66757–66766 RSC.
- C. Chen, H. Xu, Y. C. Qian and X. J. Huang, RSC Adv., 2015, 5, 15909–15915 RSC.
- T. Vokata and J. H. Moon, Macromolecules, 2013, 46, 1253–1259 CrossRef CAS PubMed.
- R. Sakai, A. Nagai, Y. Tago, S. Sato, Y. Nishimura, T. Arai, T. Satoh and T. Kakuchi, Macromolecules, 2012, 45, 4122–4127 CrossRef CAS.
- D. Wang and T. Michinobu, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 72–81 CrossRef CAS.
- Z. U. Levi and T. D. Tilley, J. Am. Chem. Soc., 2009, 131, 2796–2797 CrossRef CAS PubMed.
- T. Kawase, T. Fujiwara, C. Kitamura, A. Konishi, Y. Hirao, K. Matsumoto, H. Kurata, T. Kubo, S. Shinamura and H. Mori, Angew. Chem., Int. Ed., 2010, 49, 7894–7898 CrossRef.
- A. Konishi, T. Fujiwara, N. Ogawa, Y. Hirao, K. Matsumoto, H. Kurata, T. Kubo, C. Kitama and T. Kawase, Chem. Lett., 2010, 39, 300–301 CrossRef CAS.
- D. Wang, X. K. Wang, Y. Z. Zhao, H. Gao, Y. Xing and H. Yang, Polym. Chem., 2012, 3, 914–919 RSC.
- T. Michinobu, J. Am. Chem. Soc., 2008, 130, 14074–14075 CrossRef CAS PubMed.
- T. Michinobu, H. Kumazawa, K. Noguchi and K. Shigehara, Macromolecules, 2009, 42, 5903–5905 CrossRef CAS.
- Y. R. Li and T. Michinobu, J. Mater. Chem., 2012, 22, 9513–9521 RSC.
- T. Yamamoto, T. Morikita, T. Maruyama, K. Kubota and M. Katada, Macromolecules, 1997, 30, 5390–5396 CrossRef CAS.
- L. Huo, J. Hou, H. Y. Chen, S. Zhang, Y. Jiang, T. L. Chen and Y. Yang, Macromolecules, 2009, 42, 6564–6571 CrossRef CAS.
- M. Sheik-Bahae, A. A. Said, T. H. Wei, D. J. Hagan and E. W. van Stryland, IEEE J. Quantum Electron., 1990, 26, 760–769 CrossRef CAS.
- X. Liu, D. Wang, H. Gao, Z. Yang, Y. Xing, H. Cao, W. L. He, H. H. Wang, J. M. Gu and H. Y. Hu, Phys. Chem. Chem. Phys., 2016, 18, 7341–7348 RSC.
- Y. S. Mi, P. X. Liang, Z. Yang, D. Wang, H. Cao, W. L. He, H. Yang and L. Yu, ChemistryOpen, 2016, 5, 71–77 CrossRef CAS PubMed.
- D. Wang, Q. S. Guo, H. Gao, Z. Yang, Y. Xing, H. Cao, W. L. He, H. H. Wang, J. M. Gu and H. Y. Hu, Polym. Chem., 2016, 7, 3714–3721 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra05000j |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.