DOI:
10.1039/C6RA04609F
(Paper)
RSC Adv., 2016,
6, 43663-43671
Novel hemocompatible nanocomposite hydrogels crosslinked with methacrylated gelatin
Received
21st February 2016
, Accepted 17th April 2016
First published on 19th April 2016
Abstract
Methacrylated gelatin (MA-gelatin) is a gelatin derivative synthesized by the reaction with methacrylic anhydride; the degree of substitution (DS) of primary amines by methacrylamide groups in the gelatin is closely related to the dosage of methacrylic anhydride. In this work, MA-gelatin has been developed as a macromolecular crosslinker to prepare novel nanocomposite hydrogels (NC gels) based on polyacrylamide (PAAm) and LAPONITE® nanoclay. Compared with unmodified gelatin, MA-gelatin improves the stability of the LAPONITE® suspension in the pre-gel system. FTIR results confirm the success of gelatin modification, in agreement with the 1H NMR result, and the copolymerization of MA-gelatin and acrylamide monomers in the NC gel network. The increasing DS of MA-gelatin thus reduces the equilibrium swelling ratio (ESR) and pore sizes, and enhances the mechanical properties of the NC gels, due to the macromolecular crosslinking effect. Unlike the small molecular crosslinker N,N′-methylenebisacrylamide (BIS), MA-gelatin shows a stronger influence on the swelling behaviours and mechanical properties of NC gels. Moreover, the MA-gelatin crosslinked NC gels exhibit decreased bovine serum albumin (BSA) adsorption, prolonged blood-clotting time and nonhemolytic nature, indicating the improved antithrombogenicity. The results show that MA-gelatin can be a hemocompatible macromolecular crosslinker for the fabrication of biomedical materials.
1 Introduction
Hydrogels, as three-dimensional hydrophilic polymer networks, have been widely applied in contact lenses,1 artificial tendons,2 tissue engineering materials3,4 and drug delivery5 because of their high softness and moistness, similar to human tissues. Nevertheless, the application of conventional hydrogels has been limited by their uncontrollable structure, poor stimulus responsiveness and mechanical properties. In 2002, Haraguchi et al. reported a novel nanocomposite hydrogel (NC gel) which was prepared by in situ radical polymerization of N-isopropylacrylamide (NIPA) monomer in a suspension of the nanoclay LAPONITE® (layer size = 25–30 nm Φ × 1 nm).6 Unlike other inorganic clays, the LAPONITE® served as a multifunctional crosslinker, and the polymer chains were attached to the surface of clay platelets in the NC gel. The organic/inorganic network structure of the NC gel was thus formed by the combination of covalent and noncovalent interactions, which endowed the NC gel with good mechanical properties and high transparency. So far, NC gels have been intensively studied for their excellent properties and convenient preparation methods. In addition, the formation mechanism,7,8 composition optimization,6,9 morphologies10 and network structure11 have also been explored.
However, all the NC gels which have been reported have mainly been based on the monomers of acrylamide derivatives. For biomedical applications, such as controlled cell adhesion surfaces and blood contacting materials, biocompatibility is one of the most important and indispensable components. Haraguchi et al. have studied cell cultivation and detachment on the surface of a poly(N-isopropylacrylamide) (PNIPA) NC gel, and the results showed that cell adhesion and proliferation were closely related to LAPONITE® concentration.12 In addition, the PNIPA-NC gel can adsorb large quantities of BSA. This suggested that the NC gel was capable of protein adsorption and cell culture,12 whereas it may also lead to undesirable thrombosis in biomedical uses when coming into contact with blood.13 Therefore, further modification is required to improve the antithrombogenicity of NC gels.
Gelatin is a kind of natural product from collagen and is used widely in the biomedical field due to its excellent biocompatibility, nontoxicity and low immunogenicity. It has been recognized that gelatin is able to improve cell adhesion and proliferation,14–16 and inhibit platelet adhesion and thrombosis generation.17,18 In our previous work, the introduction of gelatin into LAPONITE®-polyacrylamide (PAAm) NC gels could substantially improve the biocompatibility of NC gels.19 However, the incorporated gelatin chains would be released from the gel network during sustained exposure to an aqueous environment, since they are immobilized in the hydrogel only by noncovalent interactions. Besides this, the contents of gelatin and LAPONITE® in the NC gels were fairly limited due to the incompatibility of the gelatin polyelectrolyte and LAPONITE® clay, which may lead to intense coacervation.
Methacrylamide-modified gelatin (MA-gelatin) has been recently developed through the direct reaction of primary amines from gelatin and methacrylic anhydride (MA).20 As a gelatin derivative, MA-gelatin retains the biocompatibility of gelatin without impairing its hydrophilicity; thus, the presence of MA-gelatin can significantly improve the adhesion, multiplication and migration of cells on hydrogels, favourable for the fabrication of tissue engineering materials.21,22 More important, MA-gelatin has the possibility of chemical crosslinking by the polymerization of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds, in contrast to unmodified gelatin.23 Therefore, it can be expected that the combined processes of polymerization and crosslinking were simultaneously performed during the synthesis of MA-gelatin crosslinked hydrogels. The compatibility of gelatin and LAPONITE® can be also enhanced due to the substitution of amino groups by methacrylamide groups in the gelatin.
C bonds, in contrast to unmodified gelatin.23 Therefore, it can be expected that the combined processes of polymerization and crosslinking were simultaneously performed during the synthesis of MA-gelatin crosslinked hydrogels. The compatibility of gelatin and LAPONITE® can be also enhanced due to the substitution of amino groups by methacrylamide groups in the gelatin.
In that context, herein we add methacrylated gelatin, instead of gelatin, into the PAAm/LAPONITE® system to prepare a novel hemocompatible NC gel, in which the MA-gelatin works as a macromolecular crosslinker immobilized in the network by the copolymerization with acrylamide monomers. The synthesis, morphology, and mechanical properties of the resulting NC hydrogels have been investigated by varying the degree of substitution (DS) of MA-gelatin. The hemocompatibility of the NC gels has been evaluated by in vitro blood compatibility assays. Our aim is to prepare a novel MA-gelatin crosslinked hemocompatible NC gel with the potential for tissue engineering applications.
2 Experimental
2.1 Materials
Acrylamide (AAm, AR), gelatin (type B, BC; ∼240 Bloom, Mw ∼ 100 kDa), methacrylic anhydride (MA, AR) and ninhydrin (AR) were purchased from Aladdin Inc. The synthetic hectorite clay of sol-forming grade LAPONITE® RDS (denoted as LAPONITE® thereafter), Na0.7+[(Si8Mg5.5Li0.3)O20(OH)4]0.7−, was modified with pyrophosphate ions (P2O74−) from gel-forming grade LAPONITE® RD and kindly provided by Rockwood. N,N′-Methylenebisacrylamide (BIS, AR), ammonium persulfate (APS, AR), and N,N,N′,N′-tetramethylenediamine (TEMED, AR) were purchased from Sigma-Aldrich. A stock suspension of LAPONITE® RDS was prepared with deionized water at 5% w/v under vigorous stirring for at least 6 h, so that it was well dispersed into a complete exfoliated state (∼25 nm). The concentrations of the stock solutions of TEMED, APS and BIS were fixed at 10% v/v, 4% w/v and 0.8% w/v, respectively. They were prepared just before use.
For the present study, the gelatin B was used from one single batch. Its free amino groups were quantitatively measured by the ninhydrin assay24 with glycine solutions in various concentrations as standards (Fig. 1). According to the calibration curve, the amount of free amino groups in the gelatin was 0.075 mmol g−1, lower than the reference value.25 The isoelectric point (IEP) of the gelatin was at pH 4.9, as measured by the potentiometric titration method using a Nano ZS instrument (Malvern Co., UK).
 |
| | Fig. 1 Standard curve of ninhydrin test for glycine. | |
2.2 Modification of gelatin
Methacrylated gelatin (MA-gelatin) was prepared by the reaction of gelatin with methacrylic anhydride (MA).20 In detail, 5 g gelatin (∼0.375 mmol free amino groups) was dissolved in 50 mL phosphate buffer (pH 7.5, 0.2 M) at 50 °C for 1 h. Then, 0.2–1.0 mL MA (1.30–6.49 mmol) was added dropwise while vigorously stirring for 3 h. After complete reaction, the mixture was diluted with 100 mL PBS and dialyzed against deionized water at 40 °C for 72 h to remove the methacrylic acid and other impurities. The resulting MA-gelatin was obtained as a white solid after lyophilization.
The modification of gelatin was proved by 1H-NMR spectroscopy.25,26 Samples of 50 mg gelatin or gel-MA dissolved in 550 μL of deuterium oxide (D2O) were analysed. 1H NMR spectra were collected at 35 °C and at a frequency of 600 MHz by using a Bruker AV II spectrometer. The degree of substitution (DS) is defined as the percentage of ε-amino groups that are modified and was determined by following the ninhydrin assay procedures. Briefly, the reaction of ninhydrin and free amino groups at 100 °C led to a purple compound, and the optical absorbance of the solution was recorded with UV-vis spectrophotometer at 570 nm.24 The DS of MA-gelatin was calculated using:
| |
 | (1) |
where OD
g and OD
m are the optical absorbance of gelatin and MA-gelatin, respectively.
2.3 Preparation of MA-gelatin/PAAm-LAPONITE® NC gels
The NC gels were synthesized through the free radical copolymerization of MA-gelatin and the monomer (AAm) in the dispersion of LAPONITE®. The molar ratio between acrylamide, accelerator (TEMED) and initiator (APS) was kept at 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.735:0.426. The initial concentrations of AAm and MA-gelatin were respectively set at 10% w/v and 2% w/v, while the concentration of LAPONITE® was varied from 0 to 4% w/v. LAPONITE® dispersion (0–4 mL), BIS (0–280 μL) and AAm (1.0 g) were added into a 20 mL glass tube, then 5% w/v of MA-gelatin solution (4 mL) was added, and the total volume was made up to 9.74 mL with deionized water. After placing in a water bath at 50 °C for 2 h to avoid the gelation of the mixture, the solution was bubbled with nitrogen gas for 10 min, and then APS (200 μL) and TEMED (60 μL) were added. The free radical copolymerization was carried out at 25 °C for 24 h. The as-prepared hydrogel was used for swelling and mechanical properties tests directly. For structural analysis, the resulting hydrogels were dialyzed against deionized water for 72 h to remove residual monomers.
:0.735:0.426. The initial concentrations of AAm and MA-gelatin were respectively set at 10% w/v and 2% w/v, while the concentration of LAPONITE® was varied from 0 to 4% w/v. LAPONITE® dispersion (0–4 mL), BIS (0–280 μL) and AAm (1.0 g) were added into a 20 mL glass tube, then 5% w/v of MA-gelatin solution (4 mL) was added, and the total volume was made up to 9.74 mL with deionized water. After placing in a water bath at 50 °C for 2 h to avoid the gelation of the mixture, the solution was bubbled with nitrogen gas for 10 min, and then APS (200 μL) and TEMED (60 μL) were added. The free radical copolymerization was carried out at 25 °C for 24 h. The as-prepared hydrogel was used for swelling and mechanical properties tests directly. For structural analysis, the resulting hydrogels were dialyzed against deionized water for 72 h to remove residual monomers.
The synthesized MA-gelatin NC gels in this study are denoted as LxMyBz-DSp, where L, M, and B stand for LAPONITE®, MA-gelatin, and Bis, respectively; x, y, and z stand for 100× LAPONITE®/water (w/v), 100× MA-gelatin/water (w/v), and Bis (μL), respectively; and p represents the degree of substitution (DS) in percent. The term LxMyBz is eliminated when only DS is varied. For example, L2M2B70-DS93 means that a gel contains 2% w/v LAPONITE®, 2% w/v MA-gelatin with 93% DS, and 70 μL Bis during the synthesis process. The concentration of AAm was fixed at 10% w/v for all the gels.
2.4 Fourier transform infrared (FTIR) spectroscopy
The samples were freeze-dried at −50 °C in a lyophilizer (Christ ALPHA 1-2 LD) for 24 h. The infrared spectra were recorded using KBr pellets with a FTIR spectrophotometer (Nicolet IS 10) in the range of 800–4000 cm−1.
2.5 Scanning electron microscopy (SEM)
For SEM observation, the samples were first freeze-dried in a lyophilizer (Christ ALPHA 1-2 LD) for 24 h at −50 °C, and then imaged on a Phenom Pro desktop SEM (Phenom-World Company, China) operating at 20 kV.
2.6 Swelling experiments
Swelling experiments were performed at room temperature by immersing the as-prepared hydrogel samples in deionized water to reach the swelling equilibrium, and the swollen weight was recorded (Ws). The samples were then freeze-dried and weighed again to determine the weight of the dry gel (Wd). The equilibrium swelling ratio (ESR) was calculated using the following equation.| |
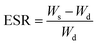 | (2) |
2.7 Measurements of mechanical properties
Compressive and tensile tests were based on the method reported in our previous work.19 All the measurements were carried out on an MTS Universal Testing Machine (CMT6202, MTS systems Co., Ltd., China) at room temperature. In the compressive test, the highest compressive strain reached a level of 80%. At least three samples were used for each test.
2.8 In vitro blood compatibility
Bovine serum albumin (BSA) adsorption. Hydrogel pieces that had been swollen in PBS for 72 h were soaked in 20 mL of BSA solution (2 mg mL−1) for 30 min, and the amount of adsorbed protein was calculated by evaluating the decrease of BSA concentration with a UV-vis spectrometer (Perkin-Elmer Lamda 25) at 277 nm.27
Blood dynamic clotting test. The anticoagulant properties of the hydrogels were evaluated by the kinetic clotting time method.28,29 0.2 mL diluted ACD blood was dropped on the surface of the samples and glass coverslips, and the clotting reaction was activated by addition of 25 μL of CaCl2 solution (0.2 mol L−1) to the ACD blood. After incubation at 37 °C for 5, 10, 20, 30, and 40 min, respectively, 50 mL deionized water was added and incubated for 10 min to lyse the red blood cells that had not been trapped in the thrombus. The concentration of free hemoglobin in the water was measured in terms of the absorbance of the supernatant at 545 nm by UV-vis spectrometer. 0.2 mL of diluted ACD blood in 50 mL of water was used as a control. The blood clotting index (BCI) was calculated as follows:| |
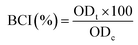 | (3) |
where ODt and ODc denote the optical density (OD) values of the test and control samples, respectively.
Hemolysis assay. The hemolytic potential of the nanocomposite hydrogels was investigated according to the reported procedure.28 The swollen hydrogel piece was immersed into a glass tube with 10 mL physiological saline and incubated for 30 min at 37 °C, and then 0.2 mL of diluted ACD blood was added into the test tube and incubated for 60 min. Positive and negative controls were prepared by adding 0.2 mL diluted ACD blood into 10 mL deionized water or physiological saline. After the incubation, all the samples were centrifuged at 2000 rpm for 5 min, and the optical density of the supernatant was determined from the absorbance at 545 nm in a UV-vis spectrometer. The percentage hemolysis was calculated according to:| |
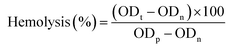 | (4) |
where ODt, ODn, and ODp denote the OD values of test, negative, and positive samples, respectively. The hemolysis results were the average of six measurements.
3 Results and discussion
3.1 Synthesis of MA-gelatin
The addition of methacrylate groups to the primary amine-containing side groups of gelatin makes it radically polymerizable (Fig. 2a).21 The successful modification of gelatin was verified by 1H NMR spectroscopy (Fig. 2b). The signal at δ = 2.8 ppm is ascribed to the lysine methylene groups in gelatin. Compared to unmodified gelatin, new signals can be observed at 5.2 ppm < δ < 5.6 ppm and at δ = 1.7 ppm in the spectrum of MA-gelatin, which are assigned to the acrylic protons and the methyl function of the introduced methacrylic groups.25,26
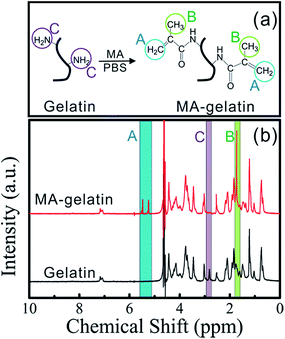 |
| | Fig. 2 (a) Schematic illustration showing the synthesis of MA-gelatin; (b) 1H NMR spectra of gelatin and MA-gelatin (DS 93%). The signals of the acrylic protons and the methyl function of the introduced methacrylic groups are denoted as A and B, while lysine methylene signal of gelatin is denoted as C. | |
Meanwhile, the signal at δ = 2.8 ppm disappears in the MA-gelatin spectrum, indicating that the amino groups of the lysine have almost entirely been substituted by methacrylamide groups in the MA-gelatin, with a high DS.
The fraction of lysine groups reacted, or the degree of substitution (DS), can be adjusted by varying the amount of MA present in the initial reaction mixture, as shown in Table 1. In fact, varying the amount of MA from 0.2 to 1.0 mL gives a 1:3.5 to 1:17.5 molar excess of MA with respect to the free amino groups of the gelatin, based on a content of 0.075 mmol of amino groups per gram of gelatin. To prepare MA-gelatin with a large number of methacrylate pendant groups (DS > 85%), the application of a much greater excess of MA is required for the conversion of the free amino groups into amide bonds.30,31 A similar result was obtained for the 93% DS at nearly 17.5-fold molar excess of MA in our study. In the subsequent synthesis of the nanocomposite hydrogels, MA-gelatin with 35%, 75% or 93% DS was used instead of pure gelatin to copolymerize with acrylamide in the LAPONITE® dispersion liquid.
Table 1 Influence of the amount of MA on DS of MA-gelatin
| Dosage of MA (mL) |
Molar ratio of free amino groups in gelatin to MA (mol mol−1) |
DS of MA-gelatin (%) |
| 0.2 |
1:3.5 |
35.4 ± 3.7 |
| 0.3 |
1:5.25 |
47.7 ± 5.1 |
| 0.4 |
1:7.0 |
75.2 ± 4.7 |
| 0.5 |
1:8.75 |
87.3 ± 2.8 |
| 1.0 |
1:17.5 |
93.4 ± 4.9 |
3.2 Stability of LAPONITE® suspension with MA-gelatin macromer
For the fabrication of NC gels with gelatin as an ionic macromer by in situ copolymerization, the difficulty is to overcome the polarization induced attractive electrostatic interaction between the negatively charged gelatin and identically charged surfaces of the LAPONITE®.32 Simple addition of pure gelatin into the LAPONITE® suspension containing monomer AAm immediately induced gelation (DS0 in Fig. 3a) leading to a steep increase in viscosity and a decrease in transmittance. It seemed that the adsorbed neutral monomer AAm was not sufficient to stabilize the sol-forming LAPONITE® in the presence of the polyampholyte gelatin, which was different to the case of small anionic monomers like sodium acrylate (SA)33 or sodium methacrylate (SMA).34 However, after heating, the gelation can be destroyed to some extent, and finally highly transparent gels are formed at limited concentrations of gelatin and LAPONITE® (≤2% w/v), as shown in Fig. 3b. In contrast, when adding modified gelatin, i.e. MA-gelatin at various DS, to the LAPONITE® under the same conditions, all of the suspensions were transparent and stable for more than 12 h. This is favorable for the in situ copolymerization. The result implies that the modification of the free amino groups of gelatin by methacrylate groups decreases the polyelectrolyte property of gelatin and weakens the electrostatic interactions between MA-gelatin and LAPONITE®. Thus, MA-gelatin can homogeneously disperse in the LAPONITE® dispersions, resulting in highly transparent ionic NC gels. The copolymerization of MA-gelatin with AAm and the role of LAPONITE® in the gel structure were then further investigated.
 |
| | Fig. 3 The stability of 2% w/v LAPONITE® suspensions containing 10% w/v AAm, 2% w/v MA-gelatin at DS 0, 35%, 75% or 93%, and 70 μL BIS. (a) Mixtures before water bath, (b) resulting hydrogels. | |
3.3 Copolymerization and structure of MA-gelatin NC gels
FT-IR spectra of gelatin before and after the modification are shown in Fig. 4. As revealed previously, the typical absorption bands around 3420 cm−1 (O–H and N–H stretching of amide A), 2928 cm−1 (C–H stretching of CH2 groups), 1645 cm−1 (amide I, peptide CO stretching), 1540 cm−1 (amide II, N–H bending coupled to C–H stretching), and 1240 cm−1 (amide III, C–N stretching and N–H bending) indicate the backbone structure of gelatin.35 The positions of these major amide bands do not change with the modification, and only the intensity of the amide I band was increased to some extent. The former implies that the methacrylation occurs in the side groups of gelatin and the latter can be due to the contribution of CC stretching (1680–1620 cm−1) in the methacrylate groups. Furthermore, in the MA-gelatin samples two IR bands at 950 cm−1 and 860 cm−1 appeared, which are characteristic of the C–H stretching of CC bonds,36 and both intensities markedly increase with increasing DS. The results further reveal that the methacrylate groups have been introduced into the structure of gelatin. The typical FT-IR spectrum of MA-gelatin NC gel is also shown in Fig. 4. Compared with MA-gelatin, the peaks at 950 cm−1 and 860 cm−1 attributed to the CC bonds disappear. In addition, the appearance of a band at ∼1000 cm−1 is typical of Si–O stretching from LAPONITE®.37 The changes indicate that the methacrylate groups in MA-gelatin have been copolymerized with AAm, resulting in crosslinked NC gels containing LAPONITE®.
 |
| | Fig. 4 FT-IR spectra of (A) pure gelatin, (B) MA-gelatin with 35% DS, (C) MA-gelatin with 93% DS, and (D) MA-gelatin NC gels (L2M2B70-DS93). | |
Fig. 5 shows the cross-sectional morphologies of the MA-gelatin NC gels. It is obvious that all the gels are honeycomb-like with uniform interconnected pores, the size of which decreases on increasing the DS of MA-gelatin. Note that the initial gel compositions (i.e., clay/polymer ratio and water content) in the L2M2B70 NC gels and the subsequent freeze-drying conditions are the same. Therefore, the difference in the porous structure can only be attributed to the varied numbers of methacrylate groups in the gelatin and the resulting crosslink density. Clearly, the higher the DS, the smaller the pore size, suggesting that the porosity can be adjusted by controlling the DS of MA-gelatin or the content of the macromolecular crosslinker. In addition, macroporous materials were found to be beneficial for cell adhesion and proliferation.38–40 Here, the pore sizes of the gels are distributed in the range of 50–200 μm, implying that they are potential cell scaffold materials.
 |
| | Fig. 5 SEM micrographs of L2M2B70 NC gels with various DS. | |
The mechanism for the formation of the highly-transparent ionic NC gels is thus proposed as follows. For the pure gelatin (DS0) system, semi-interpenetrated networks (s-IPNs) resulted, in which only AAm is crosslinked (PAAm) with LAPONITE® and Bis, while gelatin is embedded in the network in its linear form. Note that the amount of Bis used in our study is much lower than that in conventional Bis crosslinked gels, showing the typical crosslinking effect of LAPONITE®.32 However, in the presence of MA-gelatin, the formation of novel IPNs comprising covalently bonded PAAm and MA-gelatin can be expected (Fig. 6). In other words, the PAAm network can be crosslinked by either LAPONITE® or MA-gelatin, or by both. It should be noted that the MA-gelatin cannot be observed undergoing self-polymerization or polymerization with LAPONITE® to form crosslinked gels in the absence of AAm, which is likely due to the low concentrations of MA-gelatin and the corresponding methacrylic groups.25 Therefore, MA-gelatin and PAAm are at least partially crosslinked by covalent bonding on a molecular scale, although the sequence of the self-polymerization of AAm and its crosslinking with MA-gelatin cannot be distinguished at present.
 |
| | Fig. 6 Schematic representation of the MA-gelatin NC gel network prepared by the copolymerization of MA-gelatin and AAm in the dispersion of LAPONITE®. | |
3.4 Swelling behaviours
Fig. 7 shows the equilibrium swelling ratios (ESRs) of L2M2B70 NC gels with different DS of the MA-gelatin. The ESR reflects the water uptake ability of hydrogels, which is related to the network structure. Overall, the ESR largely decreases after the gelatin is methacrylated, further proving that the covalent crosslinking of MA-gelatin and PAAm occurs and thus a more rigid structure results. Moreover, with increasing DS, the crosslinking density increases as well and the polymer segments between the crosslinked points are further restricted in the network. As a result, the NC gels with higher crosslinking density hold less water than those with lower crosslinking density.
 |
| | Fig. 7 Influence of DS on the ESR of L2M2B70 NC gels containing 10% w/v PAAm, 2% w/v LAPONITE®, and 2% w/v MA-gelatin at DS 0, 35%, 75% or 93%, and 70 μL BIS. | |
For further understanding of the role of MA-gelatin in the network structure, we have used MA-gelatin or Bis separately as the crosslinker without LAPONITE® involved, to prepare MA-gelatin/PAAm hydrogels by use of an equal stoichiometry of active double bonds in the copolymerization. In addition, the total contents of pure gelatin and MA-gelatin with 93% DS are kept at 2% w/v, so that the total mass of polymers in the resulting hydrogel is constant. For comparison, each of the two crosslinkers is used in half amounts in the mixture for the hydrogel preparation. The initial compositions of the hydrogels are listed in Table 2. Here, G stands for pure gelatin, and M and B mean MA-gelatin with 93% DS and BIS, respectively. Thus, the three hydrogels presumably have same degree of covalent crosslinking. Indeed, the hydrogels containing MA-gelatin crosslinking have much lower ESR than that with Bis alone as the crosslinker, as shown in Fig. 8. It is understandable that the freedom of MA-gelatin chains is greatly restricted by the formation of covalent bonds with PAAm in the G0M2B0 network, whereas if only the PAAm network is crosslinked then the gelatin chains can be very relaxed during the swelling process, leading a higher ESR in the G2M0B2 hydrogel. Hence, MA-gelatin can not only act as a macromolecular crosslinker for the hydrogel preparation, but can also contribute to the construction of the rigid structure.
Table 2 Initial composition of the MA-gelatin/PAAm composite hydrogels
| Samples |
Gelatin/% w/v |
MA-gelatin of DS93/% w/v |
AAm/% w/v |
Bis/μL |
| G0M2B0 |
0 |
2 |
10 |
0 |
| G1M1B140 |
1 |
1 |
10 |
140 |
| G2M0B280 |
2 |
0 |
10 |
280 |
 |
| | Fig. 8 Influence of MA-gelatin and Bis contents on the ESR of MA-gelatin/PAAm hydrogels. | |
3.5 Mechanical properties
Fig. 9 shows the compressive stress–strain curves of the MA-gelatin or Bis crosslinked hydrogels in the as-prepared state (Fig. 9a) and swollen state (Fig. 9b), respectively. The compressive modulus (E) is then calculated from the slope of the initial linear area (10–17%) (shown in Table 3), and the values of compressive stress at 80% (σ) are also listed in Table 3 for comparison. As discussed above, the three hydrogels have the same number of covalent crosslinking points based on stoichiometric reaction. However, the G0M2B0 hydrogel, with MA-gelatin as a single crosslinker, exhibits the highest compressive modulus, as expected, and this is more remarkable in the swollen state. However, when approaching the compressive strain of 80%, the swollen G0M2B0 hydrogel was broken (Fig. 9b), and thus no data can be shown in Table 3. The stress–strain property further reveals the effect of the macromolecular crosslinker, MA-gelatin, on improving the rigidity of the hydrogel network, but at the sacrifice of elasticity.19
 |
| | Fig. 9 Influence of MA-gelatin and Bis contents on the mechanical properties of the hydrogels: (a) as-prepared state; (b) swollen state. | |
Table 3 Compression properties of the MA-gelatin/PAAm hydrogels
| Samples |
Compressive stress at 80% (σ)/kPa |
Compression modulus (E)/kPa |
| As-prepared |
Swollen |
As-prepared |
Swollen |
| G0M2B0 |
164.4 ± 4.8 |
— |
15.5 ± 0.1 |
13.5 ± 0.3 |
| G1M1B140 |
152.5 ± 3.9 |
75.5 ± 2.3 |
13.8 ± 0.2 |
7.3 ± 0.3 |
| G2M0B280 |
128.1 ± 2.1 |
61.1 ± 1.8 |
9.7 ± 0.5 |
4.5 ± 0.3 |
After the introduction of LAPONITE®, the compressive stress–strain properties for all the gels are improved, as shown in Fig. 10 and Table 4. Even when compressed to the strain of 90%, the MA-gelatin NC gels can quickly recover to more than 90% of the initial thickness without breakage. Fig. 10d also shows that the compressive modulus increases with increasing DS, further indicating that the enhanced compressive strength is related to the crosslinking density between MA-gelatin and PAAm. Compared with typical LAPONITE®-crosslinked hydrogels,41 the MA-gelatin NC gels can sustain higher compressive strain and reasonable deformation restorability due to the chemical crosslinking and physical entanglement of the macromolecular MA-gelatin. Meanwhile, MA-gelatin NC gels have better compressive properties than other PAAm based NC gels, such as organically modified montmorillonite/PAAm NC gels,42 natural chitosan nanofibers/PAAm NC gels43 and halloysite nanotubes/PAAm NC gels.44
 |
| | Fig. 10 Compression property of MA-gelatin NC gels (L2M2B70-DS93): (a) original state; (b) compressed state; (c) recovery state; and (d) compressive stress–strain curves of L2M2B70 NC gels with varied DS. | |
Table 4 Mechanical properties of L2M2B70 NC gels
| Samples |
σ/kPa |
E/kPa |
E*/kPa |
σb/kPa |
εb/% |
| DS0 |
64.1 ± 3.1 |
10.1 ± 0.2 |
12.7 ± 0.2 |
1059.3 ± 15.1 |
1480.7 ± 9.5 |
| DS35 |
97.1 ± 2.6 |
15.4 ± 0.1 |
16.6 ± 0.3 |
226.0 ± 4.5 |
410.2 ± 6.3 |
| DS75 |
128.4 ± 2.7 |
21.8 ± 0.6 |
22.8 ± 0.3 |
147.6 ± 2.1 |
273.7 ± 4.7 |
| DS93 |
171.0 ± 4.3 |
22.4 ± 0.3 |
24.5 ± 0.7 |
134.6 ± 3.4 |
234.7 ± 4.9 |
However, on the whole the tensile properties of the NC gels decrease greatly as the DS increases (Fig. 11). The introduction of methacrylated gelatin into the PAAm-LAPONITE® gels causes a significant decrease in the elongation ratio at break (εb) and tensile stress at break (σb), though the tensile modulus (E*) at low tensile strain increases accordingly (Table 4). This is because the chemically crosslinked network with high crosslink density or low average inter-crosslink distance always tends to be ruptured on extension,9 resulting in a decrease in εb. Indeed, the dominant LAPONITE®-crosslinked NC gel, L2M2B70-DS0, has a very high εb of ∼1500%, which agrees with the results reported by Tong.32 For the present MA-gelatin NC gels, the higher εb than that of conventional hydrogels (less than 150%)45 is mainly attributed to the presence of LAPONITE®.
 |
| | Fig. 11 Tensile stress–strain curves of L2M2B70 NC gels with varied DS. | |
3.6 In vitro blood compatibility
A thrombosis or blood clot is undesirable but frequently occurs when foreign materials come into contact with blood,46 which would lead to the failure of implants.13 The intrinsic pathway of thrombosis is recognized to be initiated by the adhesion of plasma proteins, which can activate factor XII within seconds and finally provoke the substantial formation of thrombosis.47 Therefore, the in vitro blood compatibility of NC gels is generally evaluated in terms of the amount of protein (BSA) adsorption, blood clotting index and extent of hemolysis.
Fig. 12 shows the amounts of BSA adsorbed by MA-gelatin NC gels, all of which are at a very low level (less than 0.4 mg g−1),13,48 indicating good blood compatibility of the gels. As expected, the introduction of gelatin improves the wettability and hydrophilicity of the NC gel surfaces, which in turn increase the antithrombogenicity.46,49,50 Moreover, the adsorption slightly decreases with increasing DS. This can be explained by the reduction in free amino groups of MA-gelatin, which have a high affinity for platelets and protein.13,23
 |
| | Fig. 12 The amount of BSA adsorbed by L2M2B70 NC gels with varied DS. | |
The blood clotting behaviors of the hydrogels are shown in Fig. 13. A higher value of blood clotting index indicates a longer clotting time and a better antithrombogenicity.51 The results show that all the MA-gelatin NC gels have a higher BCI than glass, owing to the fact that gelatin can inhibit the platelet adhesion and thrombin generation by impairing the functionality of plasma-vWF (von Willebrand factor).17 The presence of LAPONITE® seems not to decrease the blood compatibility with its negatively charged surface.48 Note that increasing DS can improve the BCI, but the DS35 hydrogel has the highest BCI. This result may be due to the reduction of the free amino groups of gelatin and the decrease of water uptake capacity.13
 |
| | Fig. 13 Blood clotting index of L2M2B70 NC gels with varied DS and glass as reference at 37 °C. | |
Fig. 14 shows the degree of erythrolysis and hemoglobin dissociation in contact with MA-gelatin NC gels at 37 °C for 60 min. The hemolytic ratios for all the NC gels are below the national and international permissible level of 5%, indicating they are nonhemolytic.52 Clearly, the substitution of free amino groups doesn't impair the biocompatibility of gelatin.
 |
| | Fig. 14 Hemolytic extents of L2M2B70 NC gels with varied DS at 37 °C for 60 min. | |
4 Conclusions
In this article, we have introduced MA-gelatin, instead of pure gelatin, into a PAAm/LAPONITE® system to create a novel protein/PAAm-nanoclay composite hydrogel via in situ copolymerization of MA-gelatin and AAm in a dispersion of LAPONITE®. The results show that the modification of gelatin can improve the compatibility of gelatin and LAPONITE® by decreasing the polyelectrolyte property of gelatin and weakening the electrostatic interactions between them. Moreover, covalent bonding between PAAm and MA-gelatin can be formed. The resultant MA-gelatin NC gels are macroporous materials and the pore size can be adjusted by the DS of gelatin. The crosslinking action of MA-gelatin decreases the ESR of the hydrogels distinctly, and this effect is much stronger than with the small molecular crosslinker, Bis, at an equal stoichiometrical crosslink density. The novel hydrogels exhibit good compressive resistance due to the macromolecularly crosslinked rigid structure, but the tensile strength is weaker than that of dominant LAPONITE®-crosslinked gels. All the MA-gelatin NC gels have improved blood compatibility endowed by gelatin, and the substitution of its free amino groups seems to contribute to the hemocompatibility. Our studies indicate that MA-gelatin is not only a hemocompatible structural material for the fabrication of hydrogels, but also can be a promising macromolecular crosslinker for application in biomedical materials.
Acknowledgements
The financial support of the National Natural Science Foundation (NNSF) of China (21176159, 21476148) and National High-tech Research and Development Program (863 program) of China (2013AA06A306) is gratefully acknowledged.
Notes and references
- O. Wichterle and D. Lim, Nature, 1960, 185, 117–118 CrossRef.
- N. A. Peppas, Hydrogels in Medicine and Pharmacy, CRC press, Boca Raton, FL, 1987 Search PubMed.
- S. Stanton, V. Salih, G. Bentley and S. Downes, J. Mater. Sci.: Mater. Med., 1995, 6, 739–744 CrossRef.
- J. L. Drury and D. J. Mooney, Biomaterials, 2003, 24, 4337–4351 CrossRef CAS PubMed.
- T. R. Hoare and D. S. Kohane, Polymer, 2008, 49, 1993–2007 CrossRef CAS.
- K. Haraguchi and T. Takehisa, Adv. Mater., 2002, 14, 1120–1124 CrossRef CAS.
- K. Haraguchi, H.-J. Li, K. Matsuda, T. Takehisa and E. Elliott, Macromolecules, 2005, 38, 3482–3490 CrossRef CAS.
- S. Miyazaki, H. Endo, T. Karino, K. Haraguchi and M. Shibayama, Macromolecules, 2007, 40, 4287–4295 CrossRef CAS.
- K. Haraguchi and H.-J. Li, Macromolecules, 2006, 39, 1898–1905 CrossRef CAS.
- K. Haraguchi and K. Matsuda, Chem. Mater., 2005, 17, 931–934 CrossRef CAS.
- E. Loizou, P. Butler, L. Porcar and G. Schmidt, Macromolecules, 2006, 39, 1614–1619 CrossRef CAS.
- K. Haraguchi, T. Takehisa and M. Ebato, Biomacromolecules, 2006, 7, 3267–3275 CrossRef CAS PubMed.
- A. Bajpai and M. Sharma, J. Appl. Polym. Sci., 2006, 100, 599–617 CrossRef CAS.
- B. Balakrishnan, M. Mohanty, P. Umashankar and A. Jayakrishnan, Biomaterials, 2005, 26, 6335–6342 CrossRef CAS PubMed.
- W.-H. Chang, Y. Chang, P.-H. Lai and H.-W. Sung, J. Biomater. Sci., Polym. Ed., 2003, 14, 481–495 CrossRef CAS PubMed.
- T. Wang, X.-K. Zhu, X.-T. Xue and D.-Y. Wu, Carbohydr. Polym., 2012, 88, 75–83 CrossRef CAS.
- N. Tabuchi, J. De Haan, H. R. Gallandat, P. Boonstra and W. Van Oeveren, Thromb. Haemostasis, 1995, 74, 1447–1451 CAS.
- E. De Jonge, M. Levi, F. Berends, A. Van Der Ende, J. Ten Cate and C. Stoutenbeek, Thromb. Haemostasis, 1998, 79, 286–290 CAS.
- C. P. Li, C. D. Mu, W. Lin and T. Ngai, ACS Appl. Mater. Interfaces, 2015, 7, 18732–18741 CAS.
- A. I. Van Den Bulcke, B. Bogdanov, N. De Rooze, E. H. Schacht, M. Cornelissen and H. Berghmans, Biomacromolecules, 2000, 1, 31–38 CrossRef CAS PubMed.
- J. W. Nichol, S. T. Koshy, H. Bae, C. M. Hwang, S. Yamanlar and A. Khademhosseini, Biomaterials, 2010, 31, 5536–5544 CrossRef CAS PubMed.
- J. A. Benton, C. A. DeForest, V. Vivekanandan and K. S. Anseth, Tissue Eng., Part A, 2009, 15, 3221–3230 CrossRef CAS PubMed.
- D.-M. Dragusin, S. Van Vlierberghe, P. Dubruel, M. Dierick, L. Van Hoorebeke, H. A. Declercq, M. M. Cornelissen and I.-C. Stancu, Soft Matter, 2012, 8, 9589–9602 RSC.
- C. H. Yao, B. S. Liu, C. J. Chang, S. H. Hsu and Y. S. Chen, Mater. Chem. Phys., 2004, 83, 204–208 CrossRef CAS.
- E. Hoch, T. Hirth, G. E. Tovar and K. Borchers, J. Mater. Chem. B, 2013, 1, 5675–5685 RSC.
- B. H. Lee, H. Shirahama, N. J. Cho and L. P. Tan, RSC Adv., 2015, 5, 106094–106097 RSC.
- S. Jain and A. Bajpai, Des. Monomers Polym., 2013, 16, 436–446 CrossRef CAS.
- Z. X. Meng, W. Zheng, L. Li and Y. F. Zheng, Mater. Sci. Eng., C, 2010, 30, 1014–1021 CrossRef CAS.
- L. Cui, C. Tang and C. Yin, Carbohydr. Polym., 2012, 87, 2505–2511 CrossRef CAS.
- E. Hoch, C. Schuh, T. Hirth, G. M. Tovar and K. Borchers, J. Mater. Sci.: Mater. Med., 2012, 23, 2607–2617 CrossRef CAS PubMed.
- T. Billiet, B. V. Gasse, E. Gevaert, M. Cornelissen, J. C. Martins and P. Dubruel, Macromol. Biosci., 2013, 13, 1531–1545 CrossRef CAS PubMed.
- N. Pawar and H. B. Bohidar, J. Polym. Sci., Part B: Polym. Phys., 2010, 48, 555–565 CrossRef CAS.
- L. Xiong, M. Zhu, X. Hu, X. Liu and Z. Tong, Macromolecules, 2009, 42, 3811–3817 CrossRef CAS.
- X. Hu, L. Xiong, T. Wang, Z. Lin, X. Liu and Z. Tong, Polymer, 2009, 50, 1933–1938 CrossRef CAS.
- B. Li, J. F. Kennedy, Q. G. Jiang and B. J. Xie, Food Res. Int., 2006, 39, 544–549 CrossRef CAS.
- N. Moszner, J. Pavlinec, I. Lamparth, F. Zeuner and J. Angermann, Macromol. Rapid Commun., 2006, 27, 1115–1120 CrossRef CAS.
- J. Du, J. L. Zhu, R. L. Wu, S. M. Xu, Y. Tan and J. D. Wang, RSC Adv., 2015, 5, 60152–60160 RSC.
- L. Ma, C. Gao, Z. Mao, J. Zhou, J. Shen, X. Hu and C. Han, Biomaterials, 2003, 24, 4833–4841 CrossRef CAS PubMed.
- T. M. Freyman, I. V. Yannas and L. J. Gibson, Prog. Mater. Sci., 2001, 46, 273–282 CrossRef CAS.
- S. H. Oh, I. K. Park, J. M. Kim and J. H. Lee, Biomaterials, 2007, 28, 1664–1671 CrossRef CAS PubMed.
- T. Wang, D. Liu, C. X. Lian, S. D. Zheng, X. X. Liu and Z. Tong, Soft Matter, 2012, 8, 774–783 RSC.
- E. Helvacioglu, V. Adin, T. Nugay, N. Nugay, B. G. Uluocak and S. Sen, J. Polym. Res., 2011, 18, 2341–2350 CrossRef CAS.
- C. J. Zhou and Q. Q. Wu, Colloids Surf., B, 2011, 84, 155–162 CrossRef CAS PubMed.
- M. X. Liu, W. D. Li, J. H. Rong and C. R. Zhou, Colloid Polym. Sci., 2012, 290, 895–905 CAS.
- H. Kaur and P. Chatterji, Macromolecules, 1990, 23, 4868–4871 CrossRef CAS.
- K. L. Menzies and L. Jones, Optom. Vis. Sci., 2010, 87, 387–399 Search PubMed.
- M. B. Gorbet and M. V. Sefton, Biomaterials, 2004, 25, 5681–5703 CrossRef CAS PubMed.
- S. Prabhakar, J. Bajpai and A. K. Bajpai, Polym.-Plast. Technol. Eng., 2012, 51, 1443–1450 CrossRef CAS.
- K. Kataoka, H. Ito, H. Amano, Y. Nagasaki, M. Kato, T. Tsuruta, K. Suzuki, T. Okano and Y. Sakurai, J. Biomater. Sci., Polym. Ed., 1998, 9, 111–129 CrossRef CAS PubMed.
- H. Bundela and A. Bajpai, eXPRESS Polym. Lett., 2008, 2, 201–213 CrossRef CAS.
- V. Shankarraman, G. Davis-Gorman, J. G. Copeland, M. R. Caplan and P. F. McDonagh, J. Biomed. Mater. Res., Part B, 2012, 100, 230–238 CrossRef PubMed.
- C. D. Mu, F. Liu, Q. S. Cheng, H. L. Li, B. Wu, G. Z. Zhang and W. Lin, Macromol. Mater. Eng., 2010, 295, 100–107 CAS.
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 

















