
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Abnormally high oscillator strengths of the graphene nanoribbons electronic spectrum: quantum chemistry calculations
V. G.
Maslov
a,
Andrey I.
Svitenkov
*a and
V. V.
Krzhizhanovskaya
 *abc
*abc
aITMO University, Saint Petersburg, Russia. E-mail: svitenkov@yandex.ru
bSt. Petersburg Polytechnic University, Russia
cUniversity of Amsterdam, The Netherlands. E-mail: V.Krzhizhanovskaya@uva.nl
First published on 2nd August 2016
Abstract
Armchair-edged narrow graphene nanoribbons (GNRs) are modelled by semi-empirical Hartree–Fock based quantum chemistry method ZINDO/S-CI. Electronic transitions with abnormally high oscillator strengths of over 200 are found in long GNRs (over 150 hexagonal carbon rings). We argue that this high optical absorption is caused by the structure of molecular orbitals and by the system size, and not by the configuration interaction.
Introduction
Graphene-based nanostructures have been in the spotlight of attention in research and industry due to their unique mechanical, chemical and electronic properties1–4 with applications in electronics, composite materials, biological engineering, energy storage, and ultrafast photonics. Especially promising are the nanoscale devices: quantum gates,5 nanoscale transistors,6 optical modulators7,8 and infrared photodetectors.9 Of particular interest are the so-called graphene nanoribbons (GNRs), or graphene strips of a nanometre width (<10 nm).GNRs possess distinctive electronic structure and optical properties. A finite energy band gap, growing with the decreased GNR width,10 makes them an attractive material for semiconductor nanoelectronics. Experimental studies and ab initio calculations also showed that narrow graphene nanoribbons have strongly anisotropic optical properties dominated by excitonic effects that include a highly selective adsorption spectrum with respect to the light polarisation. The GNR electronic properties are influenced by the width of the ribbon, by the geometrical arrangement of carbon atoms at the edge, and by passivation of the edge atoms (for example, by hydrogen). Tuning these GNR parameters, one can control the GNRs properties important for specific applications.
Optical properties of GNRs for some typical edge structures were studied by Osella et al.11 A study by Denk et al.12 was devoted to the exciton-dominated optical response of GNR with a zigzag edge and different GNR deposition precursors. Freitag et al.13 presented a spectroscopic study of photocurrent in GNR arrays over a range of mid-IR wavelengths for wide GNRs; with the width of 130 nm versus less than 10 nm in other studies,11,12 adsorption spectrum was shifted from visible range to the IR range. In all these studies, unique GNR properties are related to an exceptionally efficient absorption of light: even a mono-atomic graphene layer can absorb a few percent of the incident radiation in the visible or IR range.
A significant number of unique optical properties of nanostructures are explained by the fact that their electronic transitions are characterised by the “giant” oscillator strength.14–16 Oscillator strength marks the number of electrons oscillating per spatial dimension during an electronic transition.30 The most important for practical applications is the dominating electronic transition in the long-wave range, where it is radiating. This is precisely the situation with the J-aggregates of molecules of certain dyes.17,18 The question of the oscillator strength of electronic transitions in GNRs is of special interest, but has not been systematically addressed yet.
In this paper, we study the existence and conditions for the emergence of electronic transitions with anomalously high values of the oscillator strength in finite-length graphene nanoribbons by the methods of computational quantum chemistry. Recent quantum chemistry calculations19–21 investigated the intermolecular interactions and the reactivity of graphene sheets. Issues related to the excited states and spectral properties of graphene were also explored,13,22 but only the relative absorption of light was reported, and no absolute values of the oscillator strength.
The oscillator strength for small samples of graphene sheets with different geometric forms (square, rectangle and triangle) were calculated by Chopra et al.23 The highest oscillator strengths were found in rectangular samples, but the highest value was only f = 1.51 since the modelled GNR patches were very short. A somewhat higher but still low oscillator strength f = 3.5 was obtained in longer GNRs.11 The limitation was obviously caused by the lack of computational power. Quantum chemistry modelling of more than one thousand atoms is a challenging computational problem. In addition to the self-consistent field simulation, it also requires extra operations for calculation of the energies and intensities of the electronic transitions (configuration interaction in the Hartree–Fock method and TDDFT in the density functional theory).
Since the oscillator strength of the electronic transitions in graphene nanoribbons increases with the ribbon length, Cocchi et al.24 characterised the intensity of the electronic transitions by the relative value f/L, where L is length of the ribbon. Due to the high complexity of the TDDFT calculations, the authors of this work considered only short ribbons of up to 40 hexagonal carbon rings (or zigzag chains), which did not allow to reveal the fundamental patterns of the oscillator strength growth in long ribbons. The highest value f/L = 0.18 Å−1 was obtained.
In our study, a computationally efficient semi-empirical quantum chemistry method was employed for calculating the electronic transitions in long narrow GNRs. We found that the oscillator strength grew with increasing nanoribbon length in essentially nonlinear way. A “giant” oscillator strength of f > 200 was obtained (f/L = 1.6 Å−1) for GNRs longer than 150 carbon rings.
Methodology
The goal of our research is investigating long GNRs, which requires modelling several thousand atoms. Applying a commonly used TDDFT ab initio method would be computationally prohibitive. We therefore decided to use the semi-empirical Hartree–Fock based quantum chemistry method ZINDO/S-CI,25 an extension of the Intermediate Neglect of Differential Overlap (INDO) method. ZINDO/S is particularly successful in predicting the optical properties of systems containing H, C, N, and O atoms. It was also validated in recent studies of the optical properties of polyaromatic hydrocarbon compounds and graphene.26,27Optical electronic transitions of all the GNRs have been computed using the software package SEMP v.2.04 developed by the authors.28 It implements the ZINDO/S-CI method and allows up to 105 atoms, and up to 4 × 105 basis functions.
Verification and validation of the SEMP code has been reported by Maslov et al.28 We have also compared the spectra predicted by SEMP with the FireFly v.8.1.1 package.29 Two samples were considered, of length Ny = 10 and 15. The calculated properties of electronic transitions are the same for both methods. The strongest electronic transition has the lowest energy. The most important parameters (energies and oscillator strengths of the strongest electronic transition) are close: the difference is less than 0.2 eV for energy and 0.5 for oscillator strength. Difference of less than 10% is considered good for this methodology.
To calculate the energies of excited states and intensities of electronic transitions, we computed interactions of 931 single-excited configurations obtained by the electronic transitions from one of the 31 highest occupied molecular orbitals (HOMO) to one of the 30 lowest unoccupied orbitals (LUMO). The dipole moment components of the transitions and oscillator strengths for 100 lowest-energy excited states were calculated.
This paper examines graphene nanoribbons with an armchair edge schematically shown in Fig. 1. The width of the ribbon was varied from 3 to 6 hexagonal carbon rings (1–2 nm) and length from 50 to 200 rings (11–43 nm). The following notation is used: GNR Nx × Ny is a nanoribbon of width Nx and length Ny. Nx is the number of carbon rings across the narrow zigzag edge; Ny is the number of carbon rings along the long armchair edge. Some researchers use a different notation,24 which we give here for the reference: width NW is the number of dimeric lines along the zigzag direction (x axis), NW = 2Nx + 1; length NL is the number of zigzag chains in the armchair direction (y axis) excluding the ends, NL = Ny − 1. In our example (Fig. 1) NW = 7 and NL = 49.
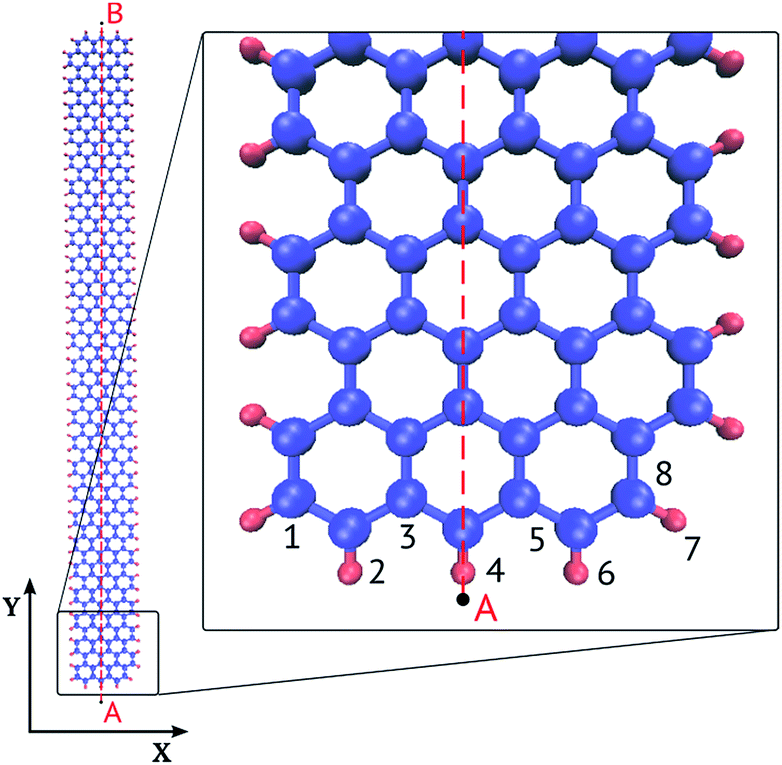 | ||
| Fig. 1 Graphene nanoribbon GNR 3 × 50 (Nx = 3, Ny = 50 carbon rings), an armchair long edge and zigzag short edge. The edge carbon atoms are passivated by hydrogen. | ||
Of fundamental importance is the following question: how significant is the influence of the configuration interaction (CI) on the excited states with high oscillator strength? To answer this question, two sets of simulation runs were performed: with and without the CI (the latter, in a single-configuration approximation), and the results were compared.
We calculated the optical absorption spectra and oscillator strengths of the electronic transitions for GNRs of different width and length. To explain the giant oscillator strength for long GNRs, we then studied spatial distributions of the orbital coefficients (weights of the contributions of the atomic orbitals to the molecular orbital) for the 2pz-orbitals of the atoms lying on line AB in Fig. 1.
In the discussion of the electronic structure of GNRs, we use the following notation: the highest occupied molecular orbital (HOMO) is designated by “0”, the lowest unoccupied molecular orbital (LUMO) by “1”, the HOMO−1 orbital by “−1”, and so on.
Results and discussion
Optical spectra and oscillator strengths
First, we calculated the optical absorption spectra and oscillator strengths of the electronic transitions for GNRs of different width and length. The oscillator strengths of the short narrow GNRs (Nx = 3, Ny ≤ 40 carbon rings) agree well with the results reported by Cocchi et al.24 The calculated spectrum of the excited states in a long GNR 3 × 120 is shown in Fig. 2. Maximum intensity is observed at the longest-wavelength transition (at E = 0.654 eV). Its oscillator strength (f = 28.4) is significantly higher than that of typical molecular systems, but it is still of the same order as in the shorter GNRs.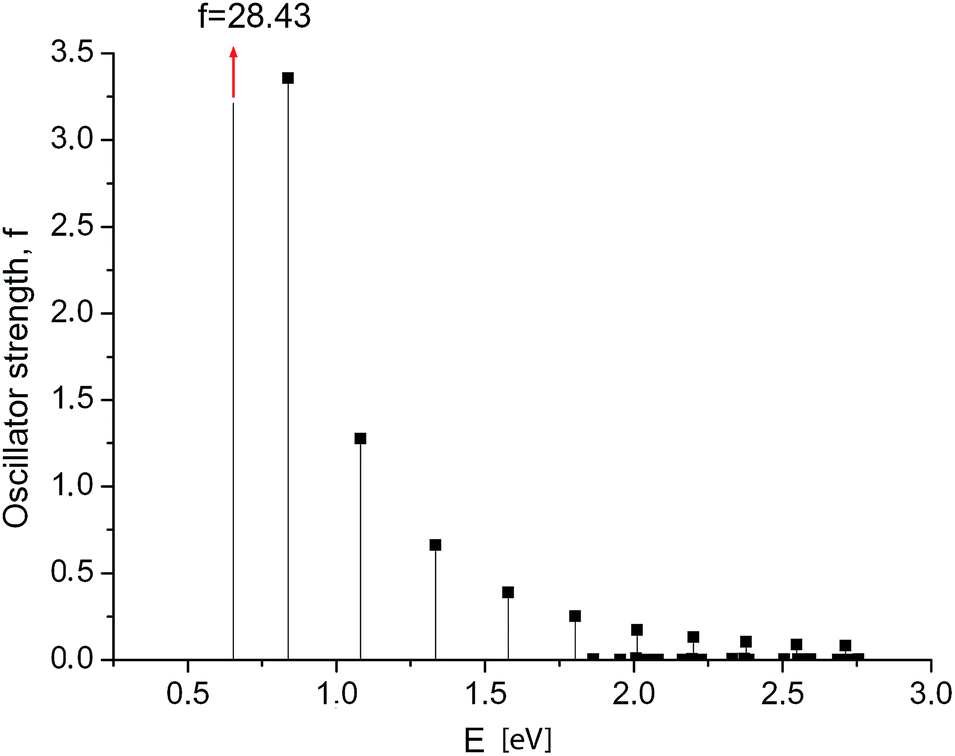 | ||
| Fig. 2 GNR 3 × 120. Spectrum of electronic transitions in graphene nanoribbon. Maximum intensity f = 28.4 is observed at the longest-wavelength transition. | ||
Information about the three most significant low-energy electronic transitions of the GNR 3 × 120 is shown in Table 1. In the longest wavelength transition #1 (at 0.654 eV), we see four decreasing contributions of single excitations: 0 → 1, −1 → 2, −2 → 3, and −3 → 4. From the remaining electronic transitions, the even ones at higher energies (#2, 4, etc.) are prohibited (i.e. they have zero intensity), while the odd ones are allowed.
| # | Taking into account configuration interactions | Single-configuration approximation | ||||
|---|---|---|---|---|---|---|
| Transition energy, eV | Oscillator strength | Configuration structure | Transition energy, eV | Oscillator strength | Configuration | |
| 1 | 0.654 | 28.4 | −0.581(0 → 1) − 0.417(−1 → 2) + 0.344(−2 → 3) − 0.286(−3 → 4) | 1.444 | 18.5 | 0 → 1 |
| 2 | 0.734 | 0.00 | 0.453(0 → 2) + 0.445(−1 → 1) − 0.277(−2 → 2) + 0.218(−3 → 3) | 1.626 | 17.1 | −1 → 2 |
| 3 | 0.838 | 3.36 | 0.418(0 → 3) + 0.324(−1 → 2) − 0.414(−2 → 1) + 0.214(−3 → 2) | 1.644 | 0.00 | 0 → 2 |
The decreasing intensity of the sequence of odd-number transitions is clearly seen in Fig. 2.
For comparison, Table 1 also shows the results of electronic transition calculations in a single-configuration approximation. In the transition #1 from the ground state to the first excited state (0 → 1), we obtained the value of the oscillator strength f = 18.5, of the same order as the result that considered the configuration interactions (CI). Hence, it follows that the high value of the oscillator strength is not particularly dependent on CI, but more on the structure involved in the transition of the molecular orbitals and on system size. With regard to the values of the transition energies, however, CI consideration is critically important for correct energy predictions.
While increasing the width of the graphene nanoribbon, we observed that dominating electronic transition is not the longest-wavelength transition anymore. For a double-width GNR 6 × 120, the highest intensity (f = 38.9) is observed at E = 2.59 eV, while the longest-wavelength transition at E = 0.27 eV has only f = 23.7 (see Fig. 3).
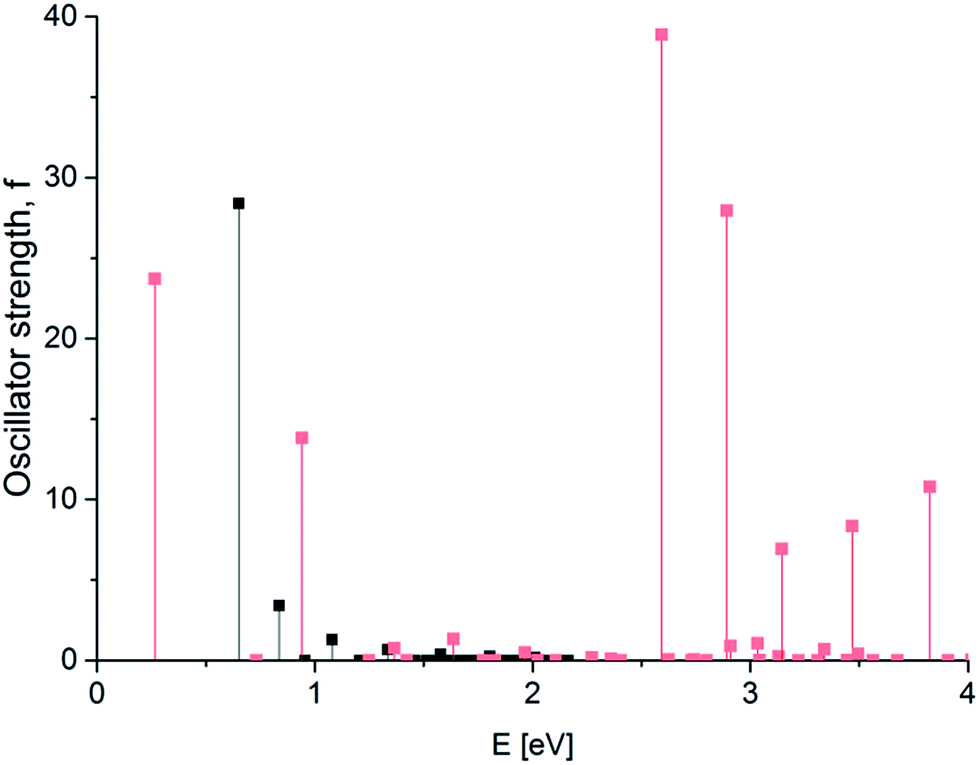 | ||
| Fig. 3 GNR 3 × 120 (black) and 6 × 120 (red). Spectrum of electronic transitions in graphene nanoribbon of increased width. | ||
Dramatic changes in the spectra of the GNR 3 × Ny were observed at Ny ≥ 150. Fig. 4 and 5 show the calculated spectra for GNR 3 × 150 and 3 × 200. As with the slightly shorter GNR 3 × 120, the spectra are dominated by the longest-wavelength transition, but the oscillator strengths are an order of magnitude higher: f = 242.2 for GNR 3 × 150 and f = 696.4 for GNR 3 × 200. A normalized oscillator strength of the longest GNR 3 × 200 is f/L = 1.6 Å−1.
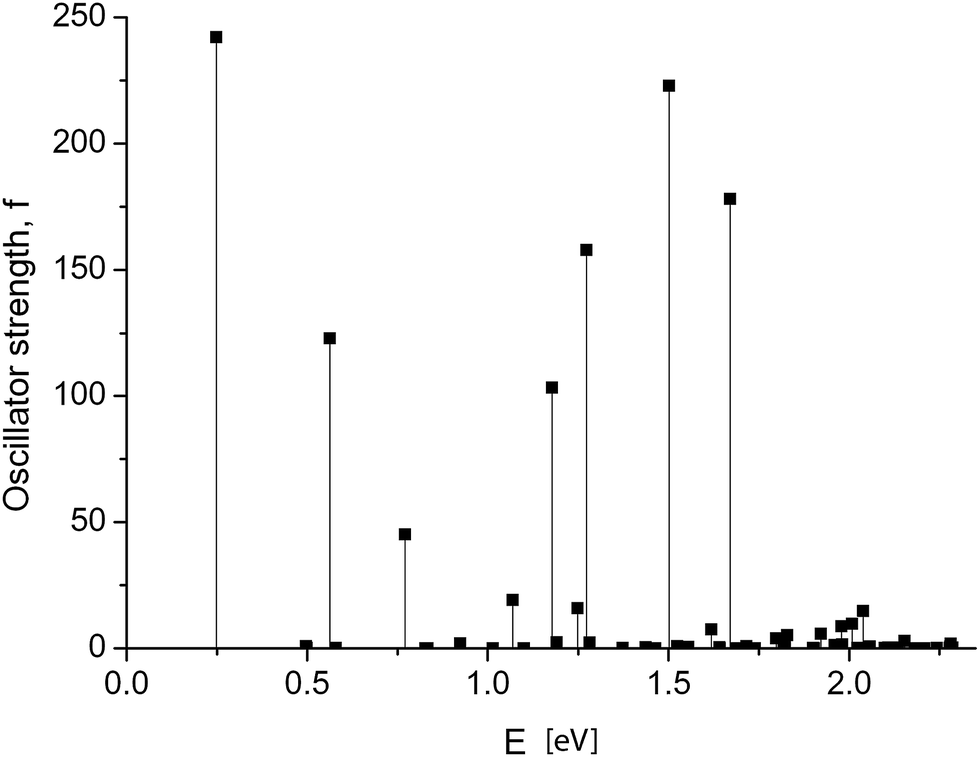 | ||
| Fig. 4 GNR 3 × 150. Spectrum of electronic transitions in graphene nanoribbon of increased length. | ||
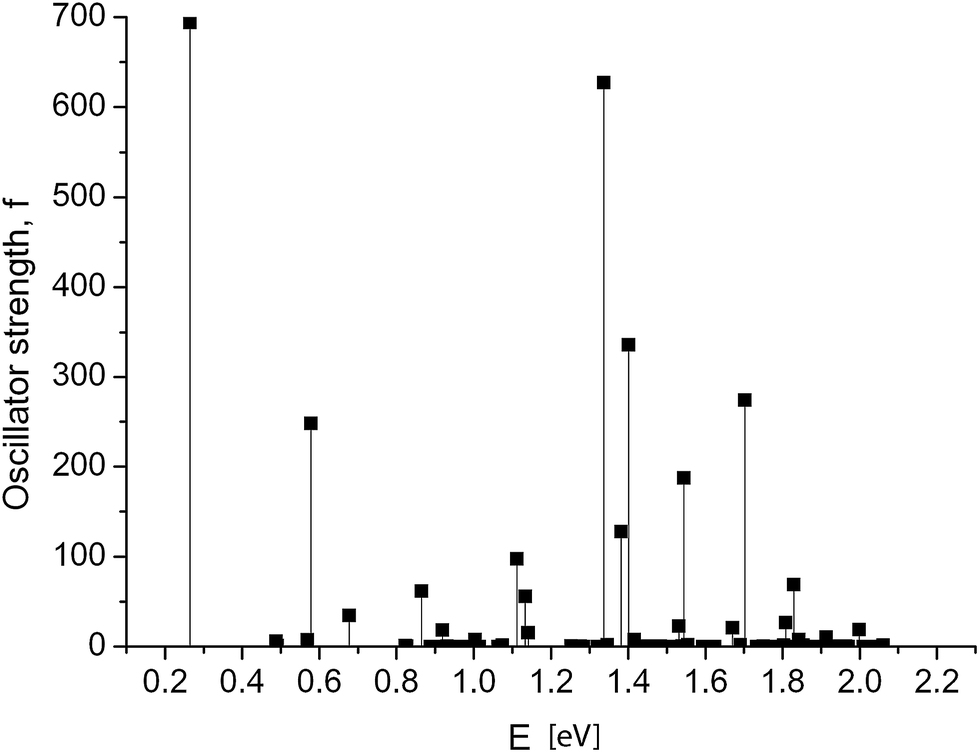 | ||
| Fig. 5 GNR 3 × 200. Spectrum of electronic transitions in graphene nanoribbon of increased length. | ||
Table 2 summarizes the data for the three lowest-energy transitions in GNR 3 × 150. For the dominating transition 0 → 1 (excitation of the ground state), the single-configuration approximation also predicts a high oscillator strength, confirming that it is mainly caused by the GNR length and by the molecular orbitals structure, and not by the configuration interactions.
| # | Taking into account configuration interactions | Single-configuration approximation | ||||
|---|---|---|---|---|---|---|
| Transition energy, eV | Oscillator strength | Configuration structure | Transition energy, eV | Oscillator strength | Configuration | |
| 1 | 0.249 | 242.2 | 0.794(0 → 1) + 0.236(−1 → 2) + 0.284(−2 → 3) + 0.204(−3 → 4) | 0.6512 | 1114.4 | 0 → 1 |
| 2 | 0.497 | 0.81 | −0.447(0 → 2) + 0.442(0 → 2) + 0.471(−1 → 1) + 0.464(−3 → 1) | 0.7207 | 14.5 | −2 → 1 |
| 3 | 0.499 | 0.12 | −0.588(0 → 3) + 0.217(0 → 5) − 0.623(−2 → 1) + 0.226(−4 → 1) | 0.7331 | 14.4 | 0 → 3 |
Electronic structure analysis: spatial distribution of atomic orbital coefficients
To explain the giant oscillator strength for long GNRs, we studied spatial distributions of the orbital coefficients (weights of the contributions of the atomic orbitals to the molecular orbital) for the 2pz-orbitals of the atoms lying on the line AB in Fig. 1. Simulation results for the GNR 3 × 120 orbital coefficients of the four most important orbitals (HOMO, HOMO+1, LUMO and LUMO+1) are shown in Fig. 6. All the orbitals that participate in the transitions in the considered energy range are purely π-orbitals.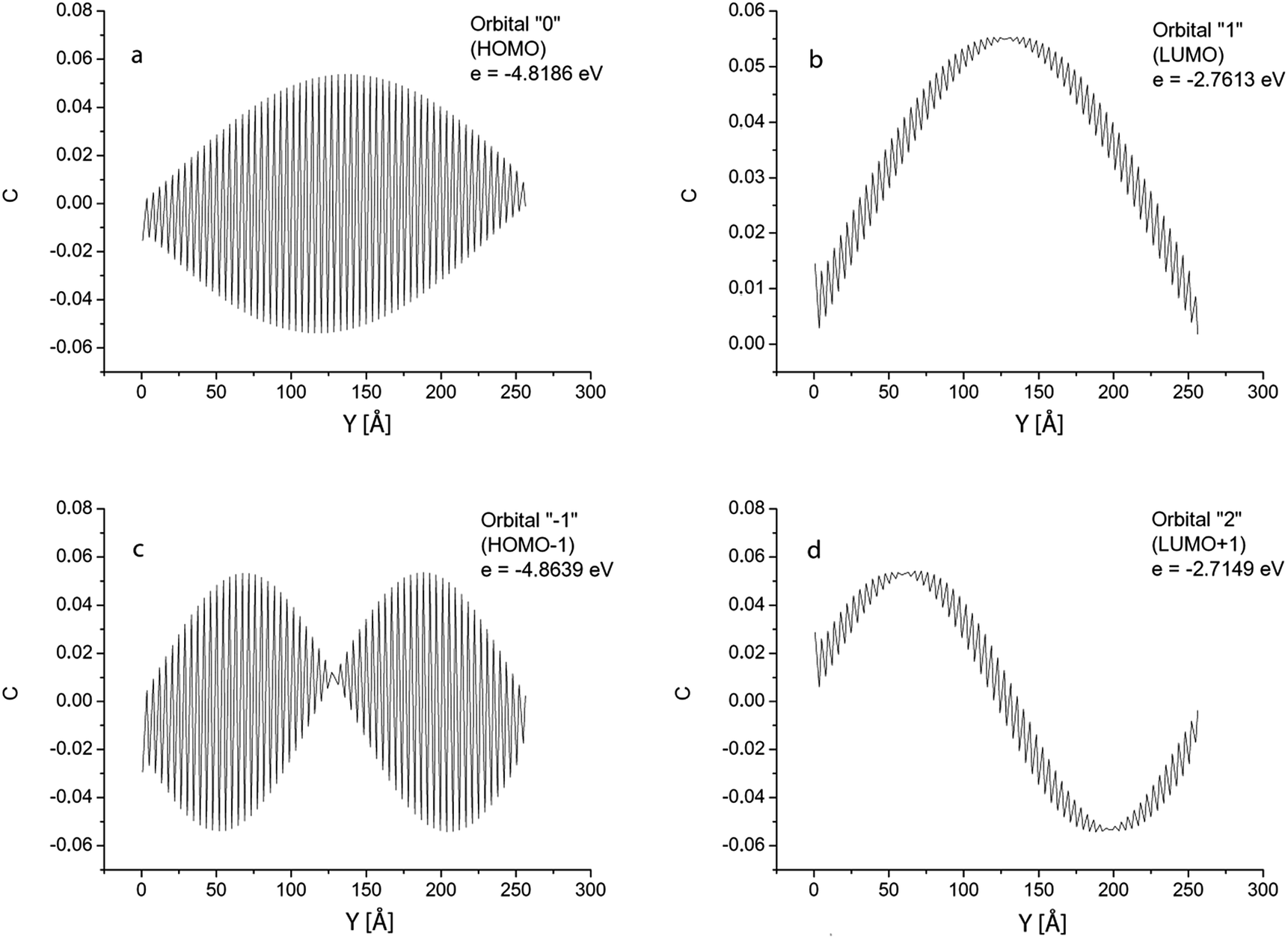 | ||
| Fig. 6 GNR 3 × 120. Spatial distribution of atomic orbital coefficients for the 2pz-orbitals of atoms lying on line AB in Fig. 1. Four most important orbitals (HOMO, HOMO−1, LUMO and LUMO+1). | ||
Orbitals 0 and −1 (HOMO and HOMO−1) have two nodal planes passing along the ribbons: one passes through atom 3, and the other through atom 6 in Fig. 1. Orbitals 1 and 2 (LUMO and LUMO+1) have five nodal planes: between the atoms 1 and 2, between 4 and 5, between 7 and 8, and in two of the above mentioned planes.
The orbital coefficients along the AB-line have a complex spatial distribution: they all experience a high-frequency variation (with a two-atom lattice period), enveloped by slow variation along the GNR length. The shape of the envelopes is determined by the number of nodal planes characterising each orbital. As can be seen in Fig. 6, there is no nodal plane in the envelope of orbital “0”; one nodal plane in the envelopes of orbitals “1” and “−1”, and 2 nodal planes for the orbital “2”.
Distributions of the GNR 3 × 150 atomic orbital coefficients corresponding to the longest-wavelength transition are shown in Fig. 7. All orbitals have five nodal planes that pass along the ribbon (the same as for the orbital “2” in GNR 3 × 120). The envelopes of the orbital coefficients bear no resemblance to any orbital envelopes in short GNRs with length Ny < 120. The minimum amplitudes of the orbitals “0” (HOMO) and “1” (LUMO) occurring with a two-atom period are close to zero (Fig. 7a and b). It is important to note that the two orbitals differ only by the symmetry: HOMO is anti-symmetric, and LUMO is symmetric with respect to the plane crossing the ribbon in the mid-length. This condition, in combination with the extended GNR length, causes a high value of the dipole moment of the excitation HOMO → LUMO transition and a high value of the corresponding oscillator strength. Orbitals “−1” and “2” (Fig. 7c and d) reveal a symmetry break with respect to the mid-length of the ribbon, but this symmetry is not “obligatory” in this case, since the system configuration is not strictly symmetric with respect to this mid-plane.
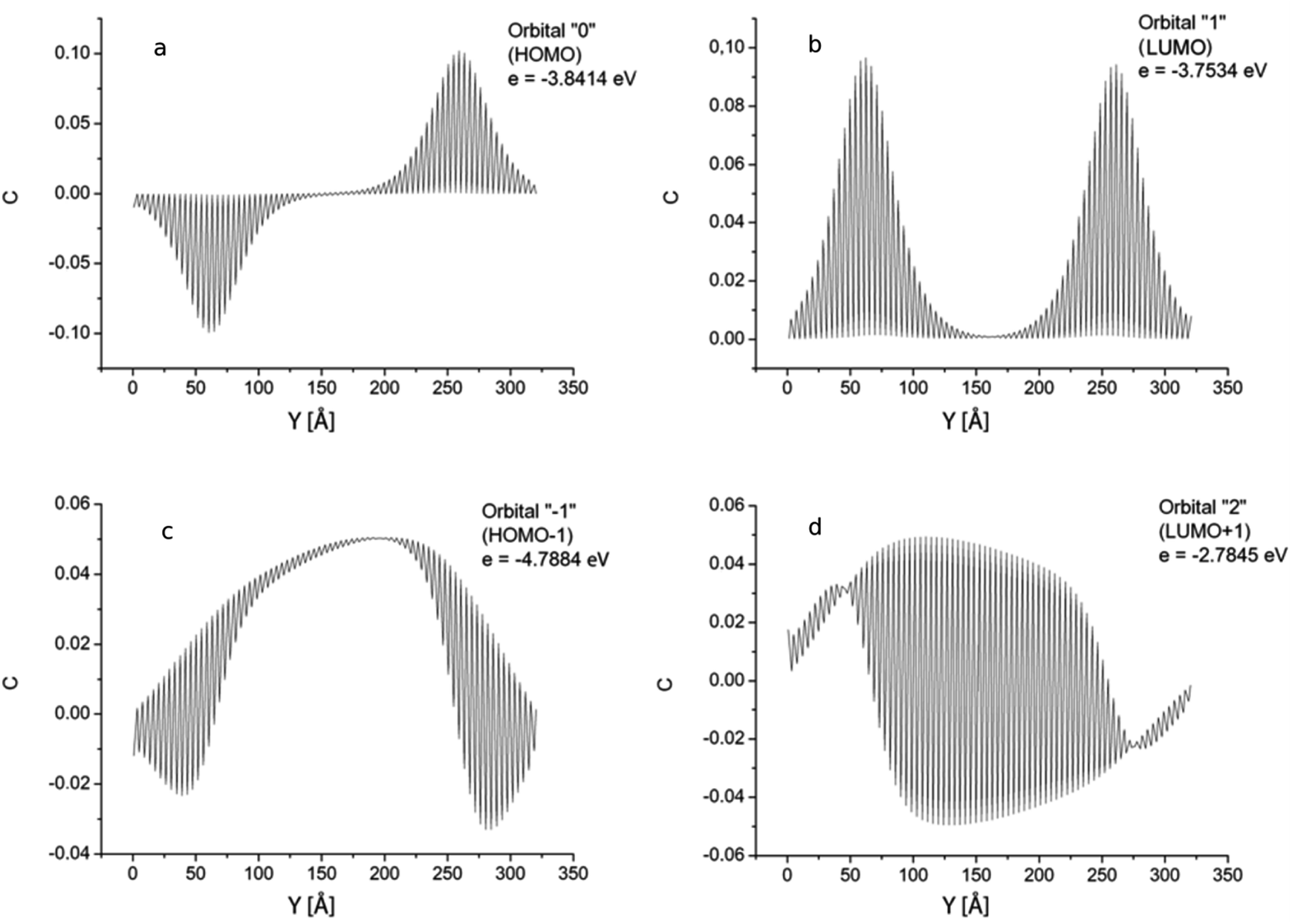 | ||
| Fig. 7 GNR 3 × 150. Spatial distribution of atomic orbital coefficients for the 2pz-orbitals of atoms lying on line AB in Fig. 1. Four most important orbitals (HOMO, HOMO−1, LUMO and LUMO+1). | ||
Curiously, the energy gap between the HOMO and LUMO orbitals in the long GNRs (Ny > 150) is considerably smaller than in short GNRs (Ny < 120). This leads to a very slow convergence of the self-consistent field calculation for large systems, which can only be achieved by using special computational procedures, for instance the level-shifting method.
Conclusions
Our quantum-chemical calculations of long narrow armchair-edged graphene nanoribbons (width Nx = 3 and 6, length Ny = 120–200 carbon rings) have shown that the long-wave electronic transitions have a very high oscillator strength (up to f = 696.4 for Ny = 200).The emergence of such high absorbance in GNRs longer than Ny ≥ 150 is explained by the electronic structure of the molecular orbitals. A dramatic change was observed in the long GNRs spatial distributions of HOMO and LUMO orbital coefficients along the ribbon length, compared to the shorter GNRs.
Calculations taking into account the configuration interactions and in single-configuration approximation revealed that such high oscillator strengths are due to the structure of molecular orbitals and size of the system, and not due to the configuration interactions.
The question of a limit transition for spectrum of infinitely long GNRs is of special interest. Other studies considered the GNRs with periodic boundary conditions.31 Our own calculations show that giant oscillator strengths disappear in this case and the resulting spectra are similar to the spectra of GNRs shorter than 120 carbon rings. We therefore assume that the ends of the ribbon play a very important role for the giant oscillator strength. This effect cannot be produced in periodic BC. This reasoning is indirectly supported by the analysis of orbital coefficients. This theoretical issue can be studied by another type of limit transition to infinite-length GNR. It could clarify the reasons of appearance of transitions with giant oscillator strength and the possibility to obtain them in real conditions. We plan to consider this question in a detailed theoretical analysis in the upcoming full-length paper.
Acknowledgements
This research is supported by the European Union's Horizon-2020 programme, grant # 671564, and by Ministry of Education and Science of the Russian Federation, Agreement #14.587.21.0024 (18.11.2015).References
- M. J. Allen, V. C. Tung and R. B. Kaner, Chem. Rev., 2010, 110, 132–145 CrossRef CAS PubMed.
- F. Bonaccorso, Z. Sun, T. Hasan and A. C. Ferrari, Nat. Photonics, 2010, 4, 611–622 CrossRef CAS.
- P. Avouris and M. Freitag, IEEE J. Sel. Top. Quantum Electron., 2014, 20, 72–83 CrossRef.
- A. N. Enyashin and A. L. Ivanovskii, Phys. Status Solidi B, 2011, 248, 1879–1883 CrossRef CAS.
- I. G. Karafyllidis, J. Comput. Sci., 2015, 11, 326–330 CrossRef.
- M. C. Lemme, T. J. Echtermeyer, M. Baus and H. Kurz, IEEE Electron Device Lett., 2007, 28, 282–284 CrossRef CAS.
- M. Liu, X. Yin, E. Ulin-Avila, B. Geng, T. Zentgraf, L. Ju, F. Wang and X. Zhang, Nature, 2011, 474, 64–67 CrossRef CAS PubMed.
- W. Li, B. Chen, C. Meng, W. Fang, Y. Xiao, X. Li, Z. Hu, Y. Xu, L. Tong, H. Wang, W. Liu, J. Bao and Y. R. Shen, Nano Lett., 2014, 14, 955–959 CrossRef CAS PubMed.
- M. Pourfath, O. Baumgartner, H. Kosina and S. Selberherr, in 2009 9th International Conference on Numerical Simulation of Optoelectronic Devices, IEEE, 2009, pp. 13–14 Search PubMed.
- M. Y. Han, B. Özyilmaz, Y. Zhang and P. Kim, Phys. Rev. Lett., 2007, 98, 206805 CrossRef PubMed.
- S. Osella, A. Narita, M. G. Schwab, Y. Hernandez, X. Feng, K. Müllen and D. Beljonne, ACS Nano, 2012, 6, 5539–5548 CrossRef CAS PubMed.
- R. Denk, M. Hohage, P. Zeppenfeld, J. Cai, C. A. Pignedoli, H. Söde, R. Fasel, X. Feng, K. Müllen, S. Wang, D. Prezzi, A. Ferretti, A. Ruini, E. Molinari and P. Ruffieux, Nat. Commun., 2014, 5, 4253 CAS.
- M. Freitag, T. Low, L. Martin-Moreno, W. Zhu, F. Guinea and P. Avouris, ACS Nano, 2014, 8, 8350–8356 CrossRef CAS PubMed.
- G. W. p't Hooft, W. A. J. A. van der Poel, L. W. Molenkamp and C. T. Foxon, Phys. Rev. B: Condens. Matter Mater. Phys., 1987, 35, 8281–8284 CrossRef.
- G. Xiong, J. Wilkinson, K. B. Ucer and R. T. Williams, J. Lumin., 2005, 112, 1–6 CrossRef CAS.
- A. Naeem, F. Masia, S. Christodoulou, I. Moreels, P. Borri and W. Langbein, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 91, 121302 CrossRef.
- J. Knoester, Int. J. Photoenergy, 2006, 2006, 1–10 Search PubMed.
- N. V. Vysotina, V. A. Malyshev, V. G. Maslov, L. A. Nesterov, N. N. Rosanov, S. V. Fedorov and A. N. Shatsev, Opt. Spectrosc., 2010, 109, 112–119 CrossRef CAS.
- A. N. Rudenko, F. J. Keil, M. I. Katsnelson and A. I. Lichtenstein, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 075422 CrossRef.
- P. A. Butrimov, O. Y. Anan'ina and A. S. Yanovskii, J. Surf. Invest.: X-Ray, Synchrotron Neutron Tech., 2010, 4, 476–479 CrossRef.
- C. Chen, J. Z. Wu, K. T. Lam, G. Hong, M. Gong, B. Zhang, Y. Lu, A. L. Antaris, S. Diao, J. Guo and H. Dai, Adv. Mater., 2015, 27, 303–309 CrossRef CAS PubMed.
- L. Yang, M. L. Cohen and S. G. Louie, Nano Lett., 2007, 7, 3112–3115 CrossRef CAS PubMed.
- S. Chopra and L. Maidich, RSC Adv., 2014, 4, 50606–50613 RSC.
- C. Cocchi, D. Prezzi, A. Ruini, E. Benassi, M. J. Caldas, S. Corni and E. Molinari, J. Phys. Chem. Lett., 2012, 3, 924–929 CrossRef CAS PubMed.
- J. Ridley and M. Zerner, Theor. Chim. Acta, 1973, 32, 111–134 CrossRef CAS.
- J. O. Oña-Ruales and Y. Ruiz-Morales, J. Phys. Chem. A, 2014, 118, 5212–5227 CrossRef PubMed.
- S. S. R. K. C. Yamijala, M. Mukhopadhyay and S. K. Pati, J. Phys. Chem. C, 2015, 119, 12079–12087 CAS.
- V. G. Maslov and A. I. Svitenkov, Electromagnetic Waves and Electronic Systems, 2013, 66, 66–73 Search PubMed.
- A. A. Granovsky, Firefly version 8.1.1, 2007, http://classic.chem.msu.su/gran/firefly/index.html Search PubMed.
- W. Kuhn, Helv. Chim. Acta, 1948, 31(6), 1780–1799 CrossRef CAS PubMed.
- L. Brey and H. A. Fertig, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73(23), 235411 CrossRef.
| This journal is © The Royal Society of Chemistry 2016 |