
Manipulating the multifunctionalities of polydopamine to prepare high-flux anti-biofouling composite nanofiltration membranes†
Abstract
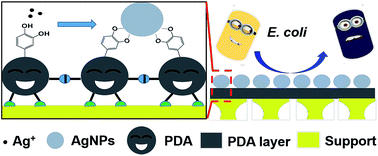
A high-flux anti-biofouling composite nanofiltration (NF) membrane was facilely prepared by simultaneously manipulating the three functionalities (adhesion, reaction and separation) of polydopamine (PDA) via a two-step dip-coating method. Stemming from the adhesion and separation functionalities, PDA was in situ deposited on a polyethersulfone (PES) substrate and formed an ultrathin dense layer, which endowed the membrane with a satisfying rejection for a broad range of small organic molecules (methyl orange of 65.65%, orange GII of 79.05%, congo red of about 98.78%, methyl blue and alcian blue of nearly 100%) and a relatively high water flux of 249.45 L m−2 h−1 MPa−1 under the operation pressure of 0.2 MPa. Stemming from the reaction functionality, PDA could reduce silver ions when exposed to AgNO3 solution and trigger the in situ generation of silver nanoparticles (AgNPs) on the membrane surface, which conferred excellent anti-biofouling properties to the membrane with low bioadhesion and high antibacterial efficiency (almost 100%) against Escherichia coli. Moreover, the adhesion and reaction functionalities have a close relation to the separation functionality of PDA. The effects of PDA deposition time, AgNO3 treatment time and AgNO3 concentration on the separation performance of the resulting composite NF membranes were systematically investigated.
 Please wait while we load your content...
Please wait while we load your content...

