
Aptasensor for the simple detection of ochratoxin A based on side-by-side assembly of gold nanorods†
Abstract
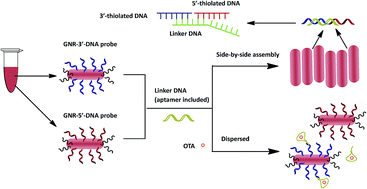
A new optical aptasensor is described based on the side-by-side assembly of gold nanorods (GNRs) for the one-step determination of ochratoxin A (OTA), which is a mycotoxin identified as a contaminant in grains and wine throughout the world. A DNA aptamer against OTA exhibiting high affinity and binding specificity has already been used as a biorecognition element to improve detection. Here, thiol-modified DNA was decorated on the side sites of GNRs acting as probes. A linker DNA containing aptamer sequences against OTA hybridized with the DNA decorated on the GNRs. It was shown that the GNRs assembled side-by-side through DNA hybridization in the absence of OTA, exhibiting strongly enhanced optical properties. However, in the presence of OTA, the GNRs dispersed as a result of specific aptamer–OTA recognition and conformational changes in the aptamer. The resulting changes in the absorption spectra of the GNRs were used for sensing. A linear correlation was demonstrated between the absorbance of the GNRs at 708 nm and the concentration of OTA over the range 0.5–20 ng mL−1. The limit of detection was 0.22 ng mL−1 (3σ), which meets the demand for detection of the allowed concentrations of OTA in foods according to European Commission Regulations (2 ng g−1). The selectivity and feasibility of the developed OTA aptasensor for the detection of OTA in red wine samples was also evaluated and it was shown to be a simple, efficient and cost-effective one-step detection method. This method is a promising tool in toxin screening to guarantee food safety and to minimize potential risks to human health.
 Please wait while we load your content...
Please wait while we load your content...

