
Propylene oxide as a dehydrating agent: potassium carbonate-catalyzed carboxylative cyclization of propylene glycol with CO2 in a polyethylene glycol/CO2 biphasic system†
Abstract
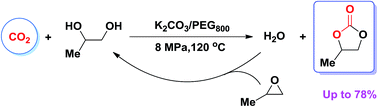
The synthesis of propylene carbonate (PC) from 1,2-propylene glycol (PG) and CO2 was smoothly performed in a PEG800 (polyethylene glycol)/CO2 biphasic system with K2CO3 as a catalyst and propylene oxide (PO) as a dehydrating agent. In the reaction of PG with CO2, PO presumably removes the water produced, and simultaneously generates more PG, both of which shift the thermodynamic control process and thus accelerate the PC synthesis. The PC yield directly from PG and CO2 reached 78% under relatively mild reaction conditions (4 MPa, 120 °C, 10 h). Notably, no additional by-product was detected in this process, resulting in economic benefits and the ease of workup procedure.
 Please wait while we load your content...
Please wait while we load your content...

