
Nanohybrids of organo-modified layered double hydroxides and polyurethanes with enhanced mechanical, damping and UV absorption properties†
Hao Zhang‡
a,
Jun Zhang‡a,
Rongping Yuna,
Zhiguo Jiang*a,
Haimei Liu*ac and
Dongpeng Yan*ab
aState Key Laboratory of Chemical Resource Engineering, Beijing University of Chemical Technology, Box 98, Beijing 100029, P. R. China. E-mail: yandongpeng001@163.com; jiangking@vip.sina.com; liuhm@shiep.edu.cn
bKey Laboratory of Theoretical and Computational Photochemistry, Ministry of Education Beijing Normal University, Beijing 100875, P. R. China
cCollege of Environmental and Chemical Engineering, Shanghai University of Electric Power, Shanghai, 200090, P. R. China
First published on 30th March 2016
Abstract
The construction of high-performance polymer–clay nanocomposites plays an important role in developing new types of organic–inorganic hybrids. In this work, a series of polymer–clay nanocomposites were prepared based on polyurethane (PU) and chemical grafting of γ-aminopropyltriethoxysilane (APS) onto dodecyl sulfonate (DS) intercalated MgAl layered double hydroxide (APS–DS–LDH). The detailed morphology, damping properties, mechanical behavior, thermal stability as well as ultraviolet absorption ability of the PU/LDH nanocomposites were studied systematically. Results reveal that the addition of APS grafted DS/LDH nanoparticles can enhance the damping properties and the thermal decomposition temperature, compared with the pristine PU. A mechanical test shows that the flexibility of the nanocomposites is significantly improved by the incorporation of APS–DS–LDH, especially for the LDH content of 3%. Meanwhile, ultraviolet absorption intensity also increases by 33%. It is expected that the PU/APS–DS–LDH nanocomposites can be used as structural damping materials and elastomers in practical applications.
1. Introduction
In recent years, the polymer–inorganic nanocomposites have attracted considerable interest because these hybrid materials display properties of both organic components (such as flexibility, easy-processing and damping ability) and inorganic units (such as high strength, chemical resistance and heat stability).1–3 To achieve an effective compatibility at the inorganic–organic interface, the high dispersion of inorganic components within the polymer matrix plays an important role. To date, several methods (such as in situ polymerization and surface modification) have been developed to obtain the adequate dispersion of the inorganic nanoparticles within the polymer matrix.4–7 For example, several previous works have been focused on the in situ preparation of polymer–clay nanocomposites by the use of cationic clays as nano-filler (e.g. montmorillonite), which gave rise to the improvement of mechanical properties.8As an important type of clays, layered double hydroxides (LDHs) stand for a large family of anionic functional materials, with the 2D geometries similar to those of layered silicates, and they also possess ultrathin layered platelets with relative high aspect ratio.9 One idealized formula of the LDHs can be expressed as [MII(1−x)MIIIx(OH)2]x+Ax/mm−·nH2O, where MII and MIII are divalent and trivalent metal cations and Am− is an exchangeable interlayer anion.10 The precursors used in LDHs preparation offer various functionalities, which allow the tuning of their properties in a wide range for applications in the fields of catalysis, hydrogenation, ion exchange, flame-retardant and biological carrier. For immobilization into polymer matrix, the LDHs nanoparticles are usually added either directly without any surface modification, or simply through a modification with a coupling agent.11 Moreover, the advantages of eco-compatible LDHs lie in their simple synthetic procedures, high degree of purity, and the ability to be assembled with a variety of organic anions12 (such as carboxylates, sulfonates, phosphates, etc.). Also, the versatility in chemical compositions and multiple interactions facilitates the optimization of dispersion degree upon hybridization within polymers.
Polyurethanes (PUs) are one of the most commonly used polymers with a wide range of physical and chemical properties since their morphology and molecular weight can be highly controlled. As a result, they are able to meet the diversified demands of modern technologies such as shape memory, coatings, adhesives, fiber, foams, rubbers, which are also known as attractive materials for sound and vibration damping application.13–15 Therefore, PUs have been considered as an excellent matrix for the preparation of different nanocomposites. In this content, extensive studies were devoted to PUs-based nanocomposites using layered silicates,16 alumina,17 carbon nanotubes18 and graphene.19 However, less reported nanocomposites have been focused on PUs/LDHs hybrids.
One of the major problems in preparing polymer–clay composite materials is the hydrophilicity of the clay that makes it less compatible with the organic polymers. Taking the LDHs clay as an example, the hydrophilic nature is attributed to the massive hydroxyl groups located at the edges of LDHs. To make the clay more compatible with polymers, the suitable organic surface modification onto LDHs is necessary, and silylation is a common and effective way to modify the physical and chemical properties of the clay. In this work, we report the preparation of PU/LDH nanocomposites using dodecyl sulfate (DS) anion and γ-aminopropyltriethoxysilane (APS) as compatibilizer. DS anion was firstly intercalated into Mg–Al–NO3 LDH via an anion-exchange process, and then the DS intercalated LDH (DS–LDH) was treated with APS, to form a bonding layer on its surface, where the pre-PU was in situ polymerized to form a new kind of polymer–clay nanocomposites (PU/APS–DS–LDH). The synthesis route and molecular illustration of nanocomposites are shown in Scheme 1. The detailed chemical structure, crystal configuration, and morphology of the nano-fillers were fully characterized by XRD, FTIR, NMR and SEM. Compared with the pristine PU, the PU/APS–DS–LDH present enhanced thermal decomposition temperature, mechanical properties, damping behaviors and ultraviolet absorption ability, which can be potentially applied in structural damping materials and related fields.
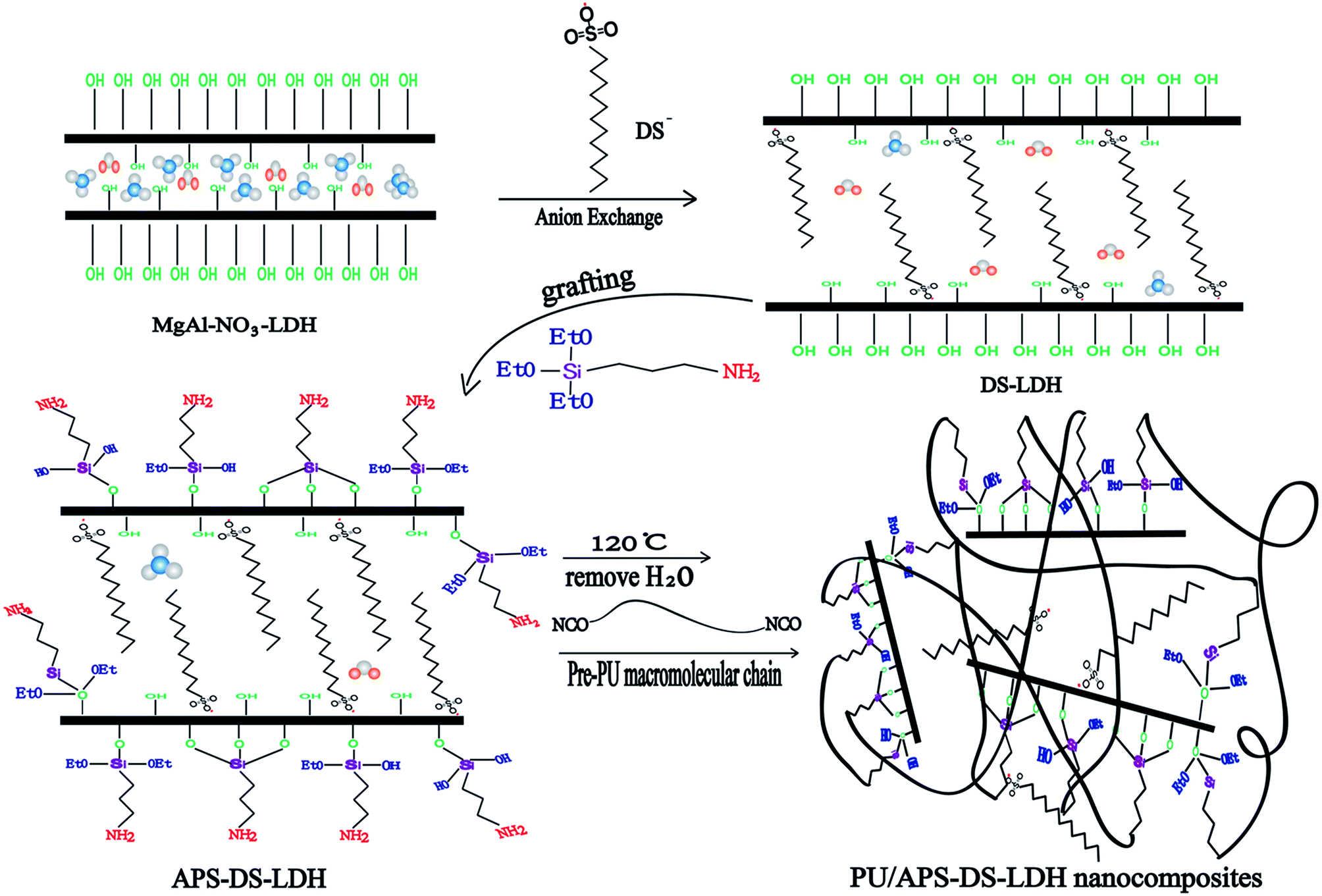 | ||
| Scheme 1 Schematic illustration of the preparation of PU/APS–DS–LDH nanocomposites. | ||
2. Experimental
2.1 Materials
PU pre-polymer was prepared from polypropylene glycol (PPG-1000) and 2,4-toluene diisocyanate (TDI), and the isocyanate content in the PU pre-polymer was 5.85%. TDI, PPG-1000, phenylmercuric acetate (C8H8HgO2) were provided by JLC (BJ) M. T. Co. Ltd. γ-aminopropyltriethoxysilane (APS), sodium dodecyl sulfate (SDS), Mg(NO3)2·6H2O, Al(NO3)3·9H2O, NaOH, trimethylolpropane (TMP), dry tetrahydrofuran (THF) were supplied by Aladdin Chemical Reagent Co. Ltd. (Shanghai, China). The deionized and decarbonated water was used in all the experimental processes.2.2 Synthesis of Mg–Al–LDH
The Mg–Al–LDH nanoparticle in nitrate form was prepared by separate nucleation and aging steps. Carbonate-free distilled water was used for dissolution of chemicals and the preparation was carried under nitrogen. 12.8 g of Mg(NO3)2·6H2O and 9.4 g of Al(NO3)3·9H2O with a Mg2+/Al3+ molar ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 were dissolved in 100 mL distilled water (solution A). 6.0 g of NaOH was dissolved in 100 mL distilled water (solution B). At room temperature, solution A and B were simultaneously flowed into the colloid mill stirring for 3 minutes with 4000 rpm, then the gel-like resultants were fast transferred into the flask, the pH value of the mixture was kept at 10. Coprecipitation was carried out under vigorous stirring at 85 °C for 12 h. The product was filtered off, washed thoroughly with distilled water and dried at 80 °C. The obtained sample was denoted as Mg–Al–NO3–LDH.
:1 were dissolved in 100 mL distilled water (solution A). 6.0 g of NaOH was dissolved in 100 mL distilled water (solution B). At room temperature, solution A and B were simultaneously flowed into the colloid mill stirring for 3 minutes with 4000 rpm, then the gel-like resultants were fast transferred into the flask, the pH value of the mixture was kept at 10. Coprecipitation was carried out under vigorous stirring at 85 °C for 12 h. The product was filtered off, washed thoroughly with distilled water and dried at 80 °C. The obtained sample was denoted as Mg–Al–NO3–LDH.
2.3 Synthesis of surfactant-intercalated Mg–Al–LDH by anionic exchange process
2.0 g of Mg–Al–NO3–LDH was dispersed into 100 cm3 distilled water (solution C). 2.72 g of SDS (0.01 mol) was dissolved in 100 mL hot distilled water (solution D). At room temperature, solution C and D were dropped into flask with vigorous stirring to exchange the anion under nitrogen. The mixture was aged at 80 °C in a water bath for about 12 h, and then the resultant slurry was filtered, washed thoroughly with distilled water, and dried at 80 °C. The obtained sample was denoted as DS–LDH.2.4 Synthesis of APS modified DS–LDH
2.0 g of DS–LDH and 5.0 mL APS (0.021 mol) were dispersed in 50 mL ethanol solution (solution E). The mixture was stirred at 80 °C for 4 h under N2. Then the product was filtered, washed thoroughly with ethanol and dried at 120 °C to remove water. The obtained sample was denoted as APS–DS–LDH.2.5 Preparation of PU/APS–DS–LDH nanocomposites by in situ polymerization
The nanocomposites of PU with the certain amount of APS–DS–LDH were prepared at room temperature by an in situ polymerization method under a nitrogen atmosphere using isocyanate terminated PU pre-polymer. 10.0 g of PU pre-polymer was dissolved into THF in a reaction flask under nitrogen. The APS–DS–LDH was dispersed in 20 mL of THF under sonication, and was subsequently added into the flask with vigorous stirring for 3 h at 60 °C. This was followed by the addition of polyol-TG (TMP and PPG-1000, –NCO:–OH = 1.2:1) as a crosslinker and two drops of phenylmercuric acetate catalyst. Stirring of the resulting solution continued for 0.5 h. The obtained crosslinked PU nanocomposites with varying APS–DS–LDH loadings (1 wt%, 2 wt%, 3 wt%, and 5 wt%) were subsequently cast on a Teflon Petri dish and kept at ambient temperature under air condition until complete removal of THF. These PU nanocomposites were denoted as PA-n where PA and n represent the elastomeric polyurethane and the weight percentage of APS–DS–LDH respectively. The neat PU elastomer was synthesized from pre-polymer in the same manner without the addition of APS–DS–LDH. PU/3 wt% MgAl–NO3–LDH and PU/3 wt% DS–LDH nanocomposites were also synthesized in the same way for comparison, and were denoted as PM3 and PD3, respectively.
2.6 Characterization
Powder XRD measurements were performed on the Rigaku XRD-6000 diffractometer, using Cu Kα (40 kV, 30 mA, λ = 0.154 nm) radiation in the 2θ range from 2 to 70° with a scan rate of 10° min−1.Fourier transform infrared (FTIR) spectra were obtained using a Vector 22 (Bruker) spectrophotometer over the wavenumber range 400–4000 cm−1 with a resolution of 4 cm−1.
Solid state 29Si CP/MAS NMR spectrum was acquired using a Bruker DSX400 NMR spectrometer operating at 79.5 MHz with a CP MAS probe and a 7 mm ZrO2 rotor. The MAS was performed at 4.5 kHz.
The morphology of APS–DS–LDH and the interfaces between LDHs and the PU matrix was investigated using scanning electron microscopy (SEM, ZEISS Supra 55) with an accelerating voltage of 20 kV.
Transmission electron microscopy (TEM) characterization was performed through a JEOL JEM-2100 electron microscope at an accelerating voltage of 200 kV.
High resolution transmission electron microscopy (HRTEM) characterization was performed through a JEOL JEM-3010 electron microscope at an accelerating voltage of 200 kV.
TG-DTA was measured using a HCT-1 thermal analysis system under ambient atmosphere with a heating rate of 10 °C min−1 from room temperature to 700 °C.
Dynamic mechanical analysis (DMA) was carried out on a DMA 1 V analyzer (Rheometric Scientific, American) using the thin film extension mode over a temperature range from −80 to 80 °C at a heating rate of 5 °C min−1 at 1 Hz.
Tensile properties were measured on an electron universal testing machine (Instron, 1185 mode, American) at a crosshead speed of 50 mm min−1. All the specimens were test three times with a dimension of 50 × 4 × 1 mm3 according to GB-T528-2009.
UV-vis absorption spectra were collected in the range from 200 to 800 nm on a Shimadzu U-3000 spectrophotometer, with a slit width of 2.0 nm.
3. Results and discussion
3.1 Characterization and morphology of APS–DS–LDH
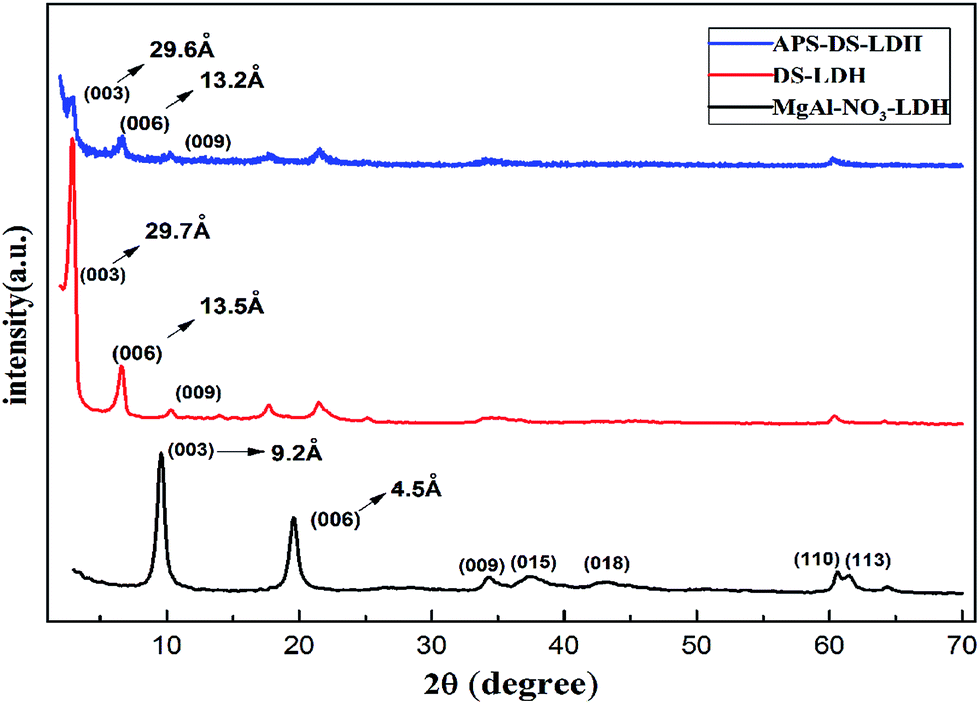 | ||
| Fig. 1 XRD profiles of MgAl–NO3–LDH, DS–LDH, and APS–DS–LDH. | ||
The diffraction of APS–DS–LDH shows that its d spacing is almost the same as that of the DS–LDH sample (Fig. 1c), that is to say, the grafting of APS onto the LDH layer does not induce an obvious change of the DS–LDH gallery. Fig. 2 shows the typical SEM image of APS–DS–LDH sample. The sample was synthesized with three steps, and presents an average diameter of about 70 nm and a shape of hexagonal platelets. HRTEM images (Fig. S1, ESI†) show the lattice fringes of MgAl–NO3–LDH, DS–LDH, APS–DS–LDH. The lattice distance is in the range of 0.15–0.16 nm, which can be attributed to the (110) plane of MgAl–LDH layers.
 | ||
| Fig. 2 Typical SEM image of APS–DS–LDH. | ||
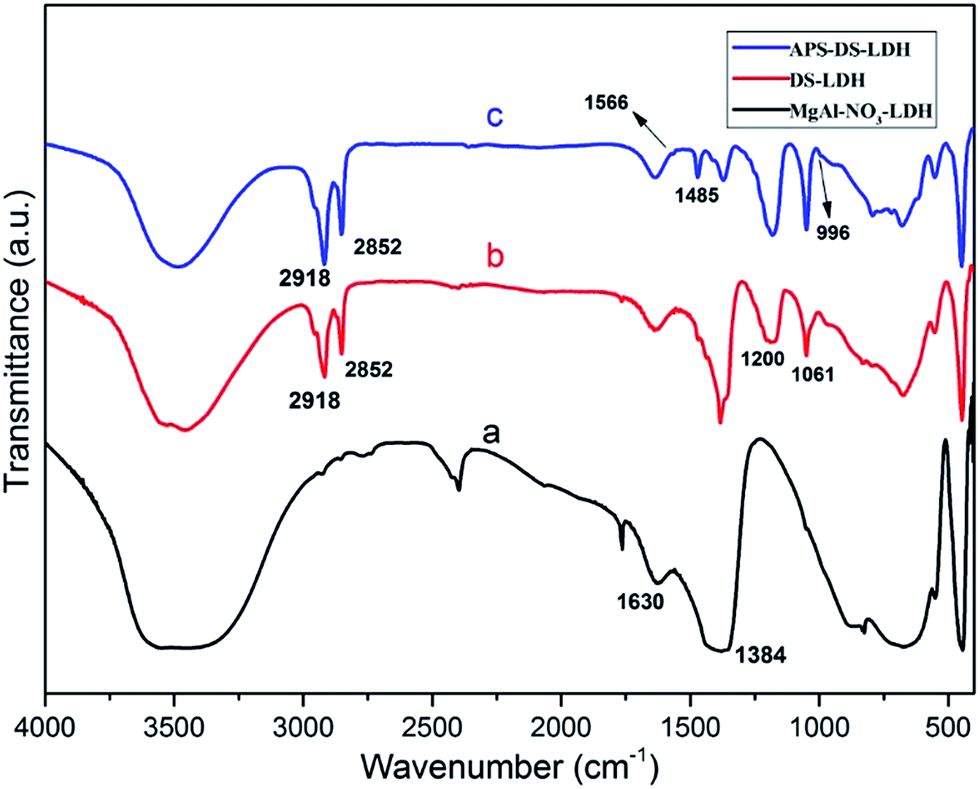 | ||
| Fig. 3 FTIR spectra: (a) MgAl–NO3–LDH; (b) DS–LDH; (c) APS–DS–LDH. | ||
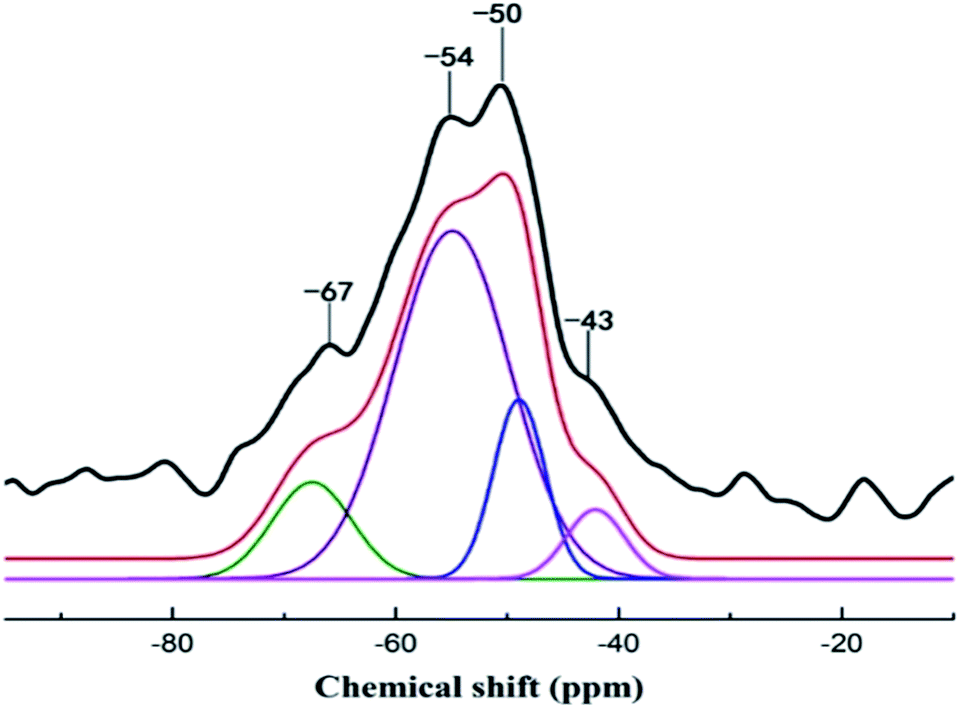 | ||
| Fig. 4 Solid state 29Si CP/MAS NMR spectrum of APS–DS–LDH. | ||
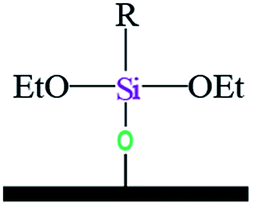
| Notation | T0 | T1 | T3 | |||
|---|---|---|---|---|---|---|
| Assignments | Silanetriol | APS | Hydrolyzed monodentate | Monodentate | Tridentate | |
| Oxane bond type | 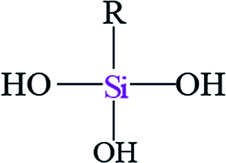 |
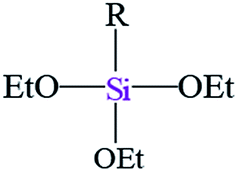 |
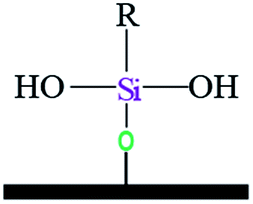 |
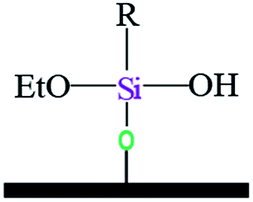 |
 |
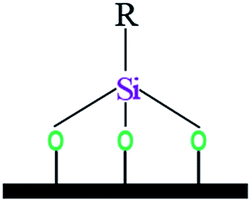 |
| Chemical shift (ppm) | −31 | −37 | −43 | −50 | −54 | −67 |
| Area (%) | 6.6 | 15.2 | 65.7 | 12.5 | ||
3.2 Morphology of PU/APS–DS–LDH nanocomposites
Fig. 5 shows the XRD patterns of the neat PU and its nanocomposites samples with different content of APS–DS–LDH loadings. APS–DS–LDH shows a sharp basal diffraction peak (003) at 2θ = 2.95° corresponding to an interlayer spacing of 2.96 nm. In addition, another diffraction peak (006) of lower intensity appears at 2θ = 6.7°. This suggests the formation of a well-stacked interlayer structure ordered arrangement of the organic anions intercalated in the interlayer galleries of nano-filler. However, the (003) basal diffraction peak of the nanocomposites (in the range from 1 wt% to 3.0 wt% LDH loading) has disappeared, indicating that the APS–DS–LDH layers have been highly dispersed and/or exfoliated in the polymer matrix due to in situ polymerization. When the LDH content increases to 5.0 wt%, the (003) reflection slightly appears, indicating that the APS–DS–LDH nanoparticles have a trend to aggregate.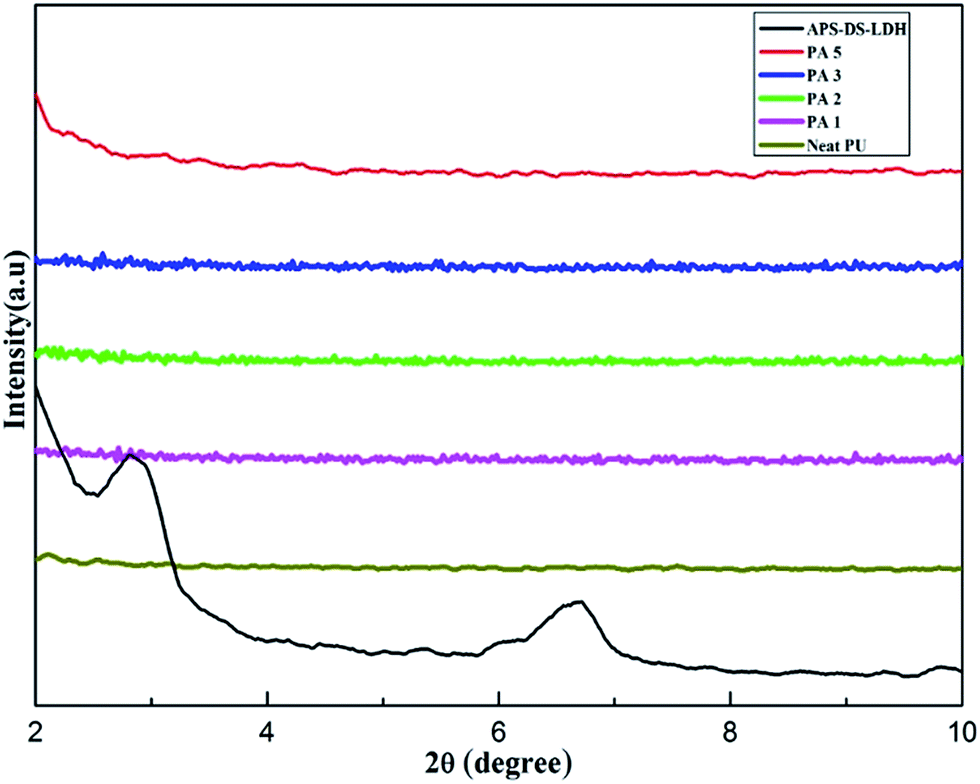 | ||
| Fig. 5 XRD patterns of pure APS–DS–LDH, neat PU and the nanocomposites. | ||
In order to further understand the dispersion degree of APS–DS–LDH in the PU matrix and compare the interfaces of PM3, PD3, PA3, SEM was also used to investigate the fresh cut surface of PU/LDH nanocomposites. SEM images show that PA3 has a better interface than PM3 and PD3 when the LDHs content is the same, suggesting that the modification of LDH by silane coupling agents plays an important role in improving the interface between LDH and PU matrix. (Fig. S2, ESI†). Fig. 6a–c show the typical SEM images of neat PU and PU/APS–DS–LDH nanocomposites containing 3 and 5 wt% APS–DS–LDH. The images shows the well dispersed hexagonal platelets in the PU matrix (Fig. 6b), suggesting that the LDH nanoparticles can serve as the new nano-filler for the PU matrix. Fig. 7 shows the TEM images of PU/APS–DS–LDH nanocomposites containing 3 wt% and 5 wt% APS–DS–LDH. The greyish white areas represent the PU matrix while the black areas represent the APS–DS–LDH layers. Fig. 7a shows that the APS–DS–LDH (3 wt%) layers are highly dispersed throughout the polymer matrix, suggesting the formation of partially exfoliated states for nanocomposites. At 5 wt% APS–DS–LDH loading, the TEM image shows the stacking of APS–DS–LDH layers in the PU matrix (Fig. 7b). These observations suggest that the polymer chains could penetrate into the interlayer region of APS–DS–LDH and result in the exfoliation of the metal hydroxide sheets apart from each other. On the other hand, the relative high concentration of APS–DS–LDH (e.g., 5 wt%) could cause the particle aggregation of LDH nano-fillers.
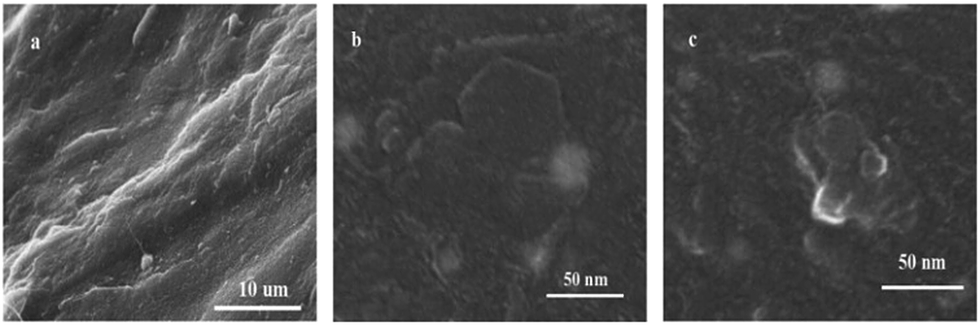 | ||
| Fig. 6 SEM micrographs of (a) neat PU, (b) PA3, (c) PA5. | ||
 | ||
| Fig. 7 TEM micrographs of (a) PA3 and (b) PA5. | ||
3.3 Dynamic mechanical analysis (DMA)
As the typical DMA curves, Fig. 8 shows the temperature dependence of storage modulus (E′), loss modulus (E′′) and loss factor (tanδ) for the PU/APS–DS–LDH nanocomposites modified with 3% APS–DS–LDH. The loss factor, tanδ, defined by the ratio E′′/E′, can be taken as a measure of the damping capability (dissipation of vibration energy) of the material.28 The glass transition temperatures (Tg) were obtained as a maximum in the tanδ curves.29 Usually, the temperature range with tanδ > 0.3 is taken as a standard to evaluate damping materials.30
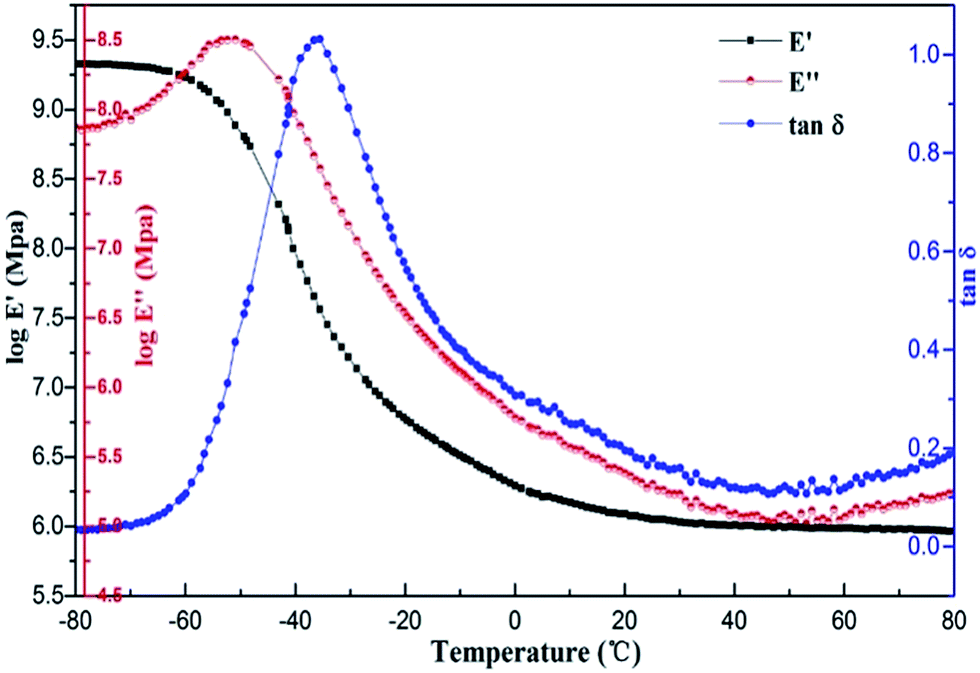 | ||
| Fig. 8 DMA curves of PU/APS–DS–LDH nanocomposites containing 3% LDH at 1 Hz. | ||
Fig. 9 and Table 2 show the damping properties of the PU/APS–DS–LDH nanocomposites at 1 Hz. Generally, glass transition temperature (Tg) can be characterized as the temperature associated with the peak magnitude of tanδ. It is well known that PU is a kind of block copolymer, which shows micro-phase separation structure for the incompatibility between soft and hard segments. Compared with that of neat PU, the Tg of soft segments (corresponding to the peak of tanδ) shifted a little to lower values and the damping temperature range (tanδ > 0.3) became wider. The lower Tg of the nanocomposites indicates that strong interaction occur between modified LDH nanoparticles and PU matrix. The interaction may further influence the original interaction between soft and hard segments of PU. As a result, the incorporation of modified LDH causes a decrease in miscibility between soft and hard segments of PU and an increase of the degree of phase separation.31–34 The temperature range with tanδ > 0.3 of neat PU was about 44.6 °C and the glass transition temperature was −34.0 °C. When the 3 wt% LDH was added, the temperature ranges with tanδ > 0.3 were approximately 52 °C, and tanδ reached a maximum value 1.04, which is improved by ca. 9.5% compared with neat PU, suggesting the enhanced damping properties upon addition of LDH nano-fillers.
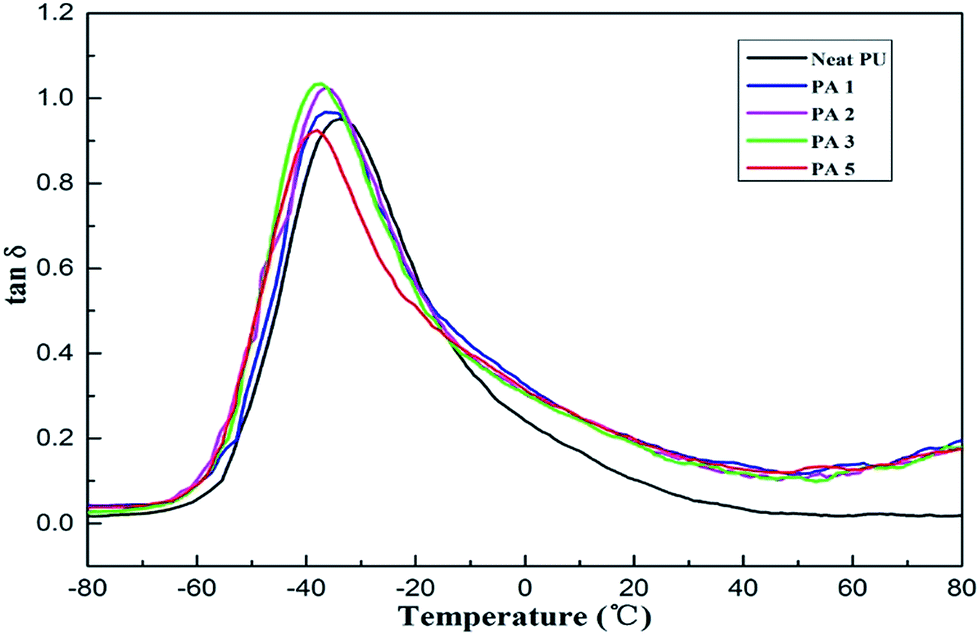 | ||
| Fig. 9 Temperature dependence of tanδ for neat PU and PU/APS–DS–LDH nanocomposites. | ||
| PU/APS–DS–LDH (%) | Temperature range (°C) (tanδ > 0.3) |
tanδ (max.) |
Tg (°C) |
|---|---|---|---|
| 0 | −50.1 to −5.5 | 0.95 | −34.0 |
| 1 | −51.1 to 2.4 | 0.96 | −35.6 |
| 2 | −52.4 to 1.0 | 1.02 | −36.6 |
| 3 | −51.5 to 1.0 | 1.04 | −37.2 |
| 5 | −52.1 to 2.3 | 0.92 | −38.0 |
The result can be attributed to that the internal friction in the nanocomposites. Damping properties are determined mainly by the energy dissipation rate in the materials.35 In this work, the internal frictions in the nanocomposites have three aspects: the internal friction of polymer chains between themselves, the internal friction between LDH nanoparticles and polymer chains, and the internal friction between LDH nanoparticles.36 When increasing the content of LDH, the internal friction between PU and LDH increases. When the LDH content reached 5 wt%, the particle aggregation tendency of LDH layers in the PU matrix increases due to the increasing filler–filler interaction over filler–polymer interaction, as also confirmed by SEM and TEM, which in turn reduces the internal friction between PU and LDH. Therefore, the 3% modified LDH/PU nanocomposite shows better damping properties. In addition, the LDH nanoparticles also had relatively high specific surface area due to their nano-scaled size and high aspect ratio that was superior to traditional filers.
3.4 Mechanical properties
The effect of formation of PU/LDH nanohybrids on their mechanical properties was studied and the results are shown in Fig. 10 and 11. It is noted in Fig. 10 that the elongation at break (EB) significantly increases by 105%, 218%, 430% and 188% for PU nanocomposites with 1, 2, 3 and 5 wt% APS–DS–LDH loadings. The maximum improvement in EB occurs at 3 wt% LDH loading, which is attributed to that the interfacial bonding between APS–DS–LDH and PU matrix makes the hybrid less susceptible to break during extension.37 Alternatively, this may be also due to the plasticization effect of the long alkyl-chain of the intercalated DS–LDH, resulting in a flexible matrix.38 However, at higher APS–DS–LDH content, the aggregated layers reduce the mobility of PU chains and shear deformation capability of PU, and thus leading to the reduction of EB.39 It is also observed from Fig. 10 that the modulus slightly decreases for all PU/APS–LDH nanocomposites compared to neat PU. This is probably due to long hydrocarbon segments of APS and DS play a key role for the improvements of the matrix flexibility over interfacial interaction between LDH and PU. These observations are similar to those in the case of thermoplastic polyolefin/SiC nanocomposites.40 Therefore, the results confirm that the EB of PU/APS–DS–LDH nanocomposites are much superior to those of neat PU.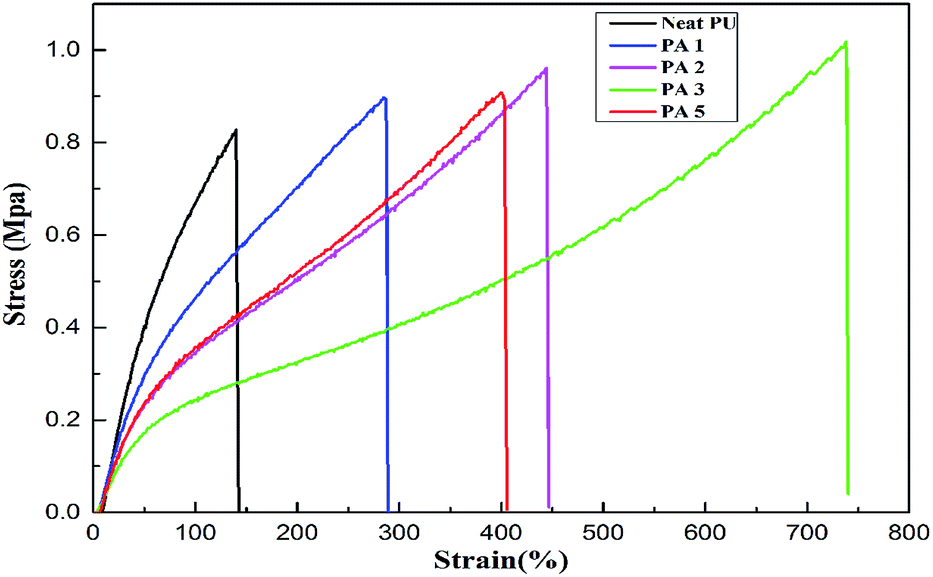 | ||
| Fig. 10 Stain–stress curves of neat PU and its nanocomposites containing different contents of APS–DS–LDH. | ||
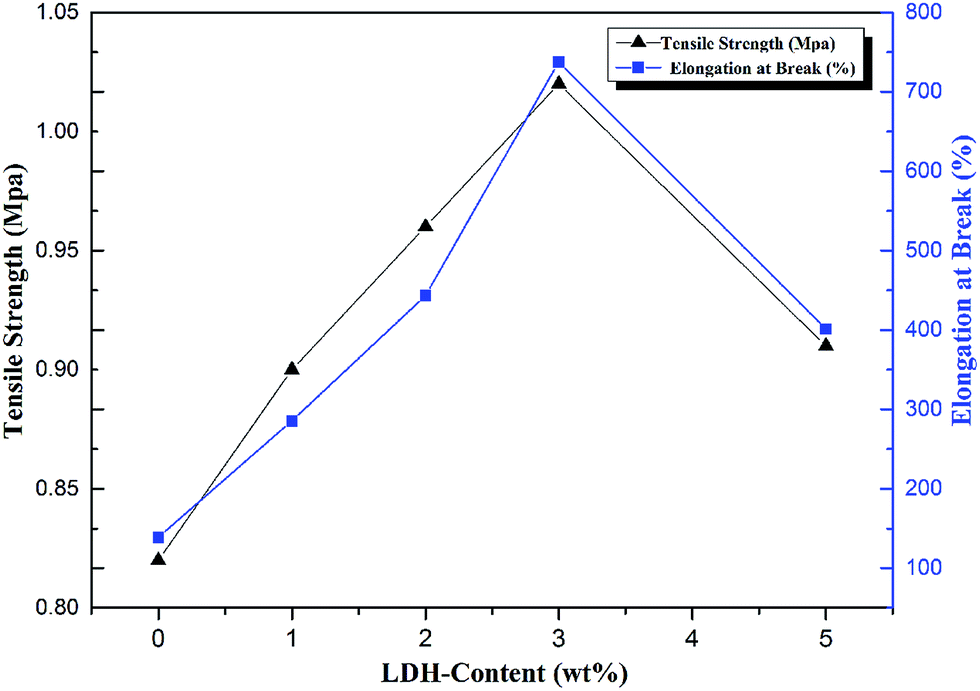 | ||
| Fig. 11 Variation of tensile strength (MPa) and elongation at break (%) of PU/APS–DS–LDH nanocomposites with different contents of APS–DS–LDH. | ||
Fig. 11 also shows that the improvements in TS for the PU/LDH nanocomposites with the optimized loading appearing at PA3. This is due to the high surface area of the APS–DS–LDH, which leads to a stronger interfacial interaction between the –OH and –NH2 groups of APS–LDH and the –NHCOO group of PU, through bridge/loop/tail linkages via hydrogen bonding forces.37 Additionally, the decrease in TS upon further addition of APS–DS–LDH in PU is attributed to the extended aggregation of APS–DS–LDH layers. Moreover, mechanical properties also have been measured on PA3, PM3 and PD3 for further comparison. PA3 shows better mechanical property compare with PM3 and PD3, further confirming the better interface between APS–DS–LDH and PU matrix (Fig. S3, ESI†).
3.5 Thermal stability of PU/APS–DS–LDH nanocomposites
The thermal degradation behaviors of PU and its nanocomposites with varying contents of APS–DS–LDH are displayed in Fig. 12 and the results are summarized in Table 3. The figure clearly demonstrates that the thermal decomposition temperature of PU nanocomposites is relatively higher than the neat PU counterpart. The degradation rate of APS–DS–LDH filled PU nanocomposites is relatively slower below 10% weight loss compared with neat PU except the filler content of 5%. This is possibly due to the dehydration of the hydroxide sheets and thermal decomposition of the alkyl chains of APS–DS–LDH before the onset of thermal decomposition of urethane linkages of PU.41 When 50% weight loss is selected as a point for comparison, the thermal decompositions for neat PU and its nanocomposites containing 1, 2, 3 and 5 wt% APS–DS–LDH are 322, 327, 339, 343 and 325 °C, respectively (Table 3). Td (50%) increases with increasing loading of APS–DS–LDH in PU and the improvement appeared maximum (21 °C) for 3 wt% filler loading in PU. This further confirms that LDH is a very effective nano-filler in improving the thermal stability as already reported in polystyrene.42 The improved behavior of PU nanocomposites is due to the relatively higher thermal stabilization effect of APS–DS–LDH. Alternatively, the homogeneous dispersion of APS–DS–LDH layers in the PU matrix provides a better barrier effect by obstructing the internal diffusion of heat and gaseous small molecules from the bulk of the PU.43 However, at higher loading (5 wt%), the decomposition temperature does not change much due to the aggregation of LDH layers and the catalytic effect in PU matrix leading to an extra heat source. It was observed that above 650 °C, very less residue (1.2 wt%) was left for neat PU, whereas 2.4, 3.6, 4.4 and 5.3 wt% of residues were left for PU nanocomposites (Table 3). The increases in residue for PU nanocomposites with increasing APS–DS–LDH might be due to the charring of the polymer, consisting of carbonaceous residue along with increasing of decomposition products of LDH (such as MgO and Al2O3).44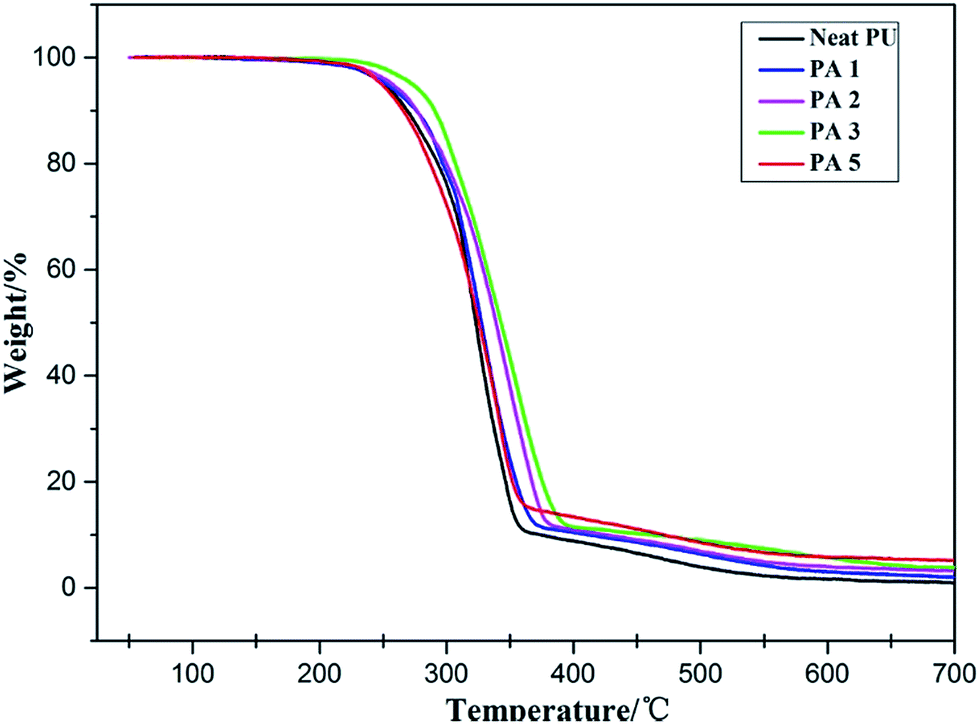 | ||
| Fig. 12 TGA curves of neat PU and its nanocomposites containing different contents of APS–DS–LDH. | ||
| PU/APS–DS–LDH (LDH%) | Td(10%) (°C) | Td(50%) (°C) | Residue at 650 (°C) |
|---|---|---|---|
| 0 | 270 | 322 | 1.25 |
| 1 | 277 | 327 | 2.42 |
| 2 | 277 | 339 | 3.55 |
| 3 | 290 | 343 | 4.40 |
| 5 | 266 | 325 | 5.33 |
3.6 UV absorption ability of PU/APS–DS–LDH nanocomposites
UV-vis absorption spectra of PU nanocomposites containing various loadings of APS–D–LDH nanoparticles are shown in Fig. 13. It can be clearly seen that a small quantity of filler notably increased the absorption by 33% when the content is 2% compared with neat PU. This observation can be related to both inherent nanoparticles ultraviolet absorption and higher probability of the light scattering.45,46 When the LDH nano-filler content continue to increase, the absorption strength decreased slightly, which is probably due to the decrease of dispersion degree. The augmented absorption in UV-vis region of PU/APS–DS–LDH nanoparticles indicates that it may also serve as promising materials for weather resistant outdoor products.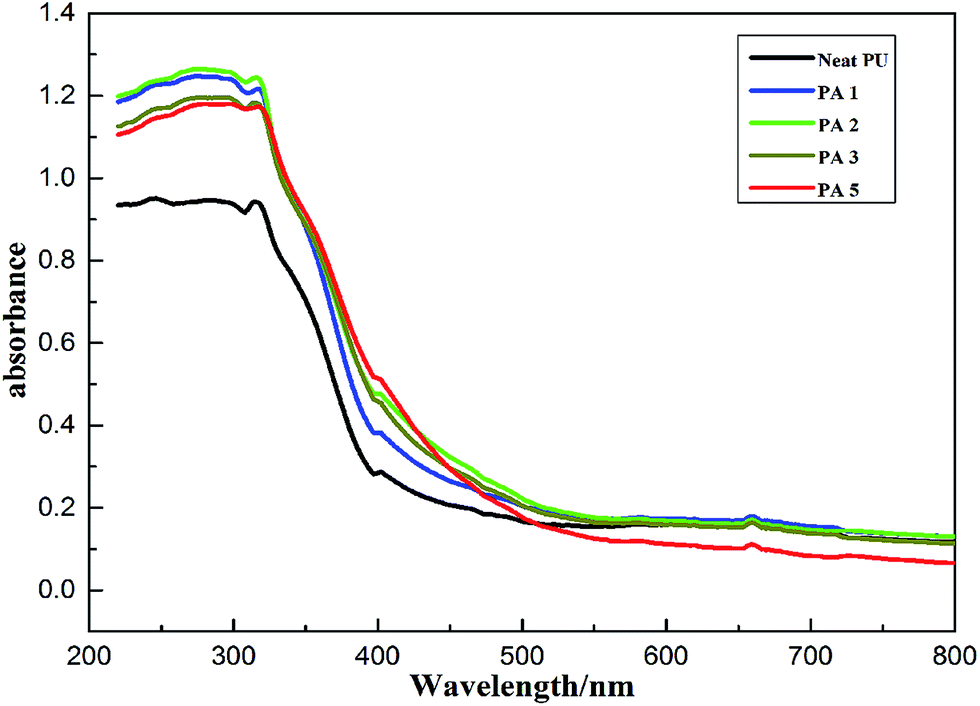 | ||
| Fig. 13 UV absorption ability of neat PU and its nanocomposites containing different contents of APS–DS–LDH. | ||
4. Conclusions
The organo-modified APS–DS–LDH has been prepared through three steps, and then PU/APS–DS–LDH nanocomposites were prepared by the in situ polymerization method. The high dispersion of nano-scaled APS–DS–LDH particles in the PU matrix has been verified by the XRD, SEM and TEM results. It has been confirmed that an effective dispersion of APS–DS–LDH results in sufficient reinforcement of their properties. For example, the elongation at break (EB) and tensile strength (TS) of the nanocomposites are significantly improved by about 430% and 24%, respectively, upon incorporation of 3.0 wt% APS–DS–LDH into PU matrix. Dynamic mechanical analysis showed the temperature ranges with tanδ > 0.3 were approximately 52 °C, and tanδ reached a maximum value 1.04 when the APS–DS–LDH content was 3%. Much improvement in thermal stability (ca. 21 °C) occurs at 3 wt% APS–DS–DH content in the PU matrix and degradation at constant weight loss shifts to longer degradation times, suggesting higher thermal stabilization of APS–DS–LDH nano-filler. In addition, the incorporation of LDH can further increase the ultraviolet absorption. Therefore, the PU/APS–DS–LDH nanocomposites can be used as multifunctional nano-filler materials in future application as PU additive. It is expected that the combination of surface modification and in situ polymerization method can be extended to other polymer–clay systems to achieve the high dispersion and compatibility at the organic–inorganic interfaces.
Acknowledgements
This work was supported by the 973 Program (Grant No. 2014CB932103), and the National Natural Science Foundation of China (NSFC), and the Beijing Municipal Natural Science Foundation (Grant No. 2152016).References
- S. Pavlidou and C. D. Papaspyrides, Prog. Polym. Sci., 2008, 33, 1119–1198 CrossRef CAS.
- M. Kotal and A. K. Bhowmick, Prog. Polym. Sci., 2015, 51, 127–187 CrossRef CAS.
- Q. Wang, J. Wu, Y. Gao, Z. Zhang, J. Wang, X. Zhang, X. Yan, A. Umar, Z. Guo and D. O'Hare, RSC Adv., 2013, 3, 26017–26024 RSC.
- S. Z. Guo, C. Zhang, H. D. Peng, W. Z. Wang and T. X. Liu, Compos. Sci. Technol., 2011, 71, 791–796 CrossRef CAS.
- F. A. He and L. M. Zhang, Compos. Sci. Technol., 2007, 67, 3226–3232 CrossRef CAS.
- Y. Z. Bao, Z. M. Huang and Z. X. Weng, J. Appl. Polym. Sci., 2006, 102, 1471–1477 CrossRef CAS.
- S. Y. A. Shin, L. C. Simon, J. B. P. Soares and G. Scholz, Polymer, 2003, 44, 5317–5321 CrossRef CAS.
- E. Manias, A. Touny, L. Wu, K. Strawhecker, B. Lu and T. C. Chung, Chem. Mater., 2001, 13, 3516–3523 CrossRef CAS.
- F. Leroux and J. P. Besse, Chem. Mater., 2001, 13, 3507–3515 CrossRef CAS.
- D. G. Evans and R. C. T. Slade, in Layered Double Hydroxides, ed. X. Duan and D. G. Evans, 2006, vol. 119, pp. 1–87 Search PubMed.
- D. Basu, A. Das, K. W. Stoeckelhuber, U. Wagenknecht and G. Heinrich, Prog. Polym. Sci., 2014, 39, 594–626 CrossRef CAS.
- D. Yan, Y. Zhao, M. Wei, R. Liang, J. Lu, D. G. Evans and X. Duan, RSC Adv., 2013, 3, 4303–4310 RSC.
- D. K. Chattopadhyay and K. Raju, Prog. Polym. Sci., 2007, 32, 352–418 CrossRef CAS.
- J. W. Cho, J. W. Kim, Y. C. Jung and N. S. Goo, Macromol. Rapid Commun., 2005, 26, 412–416 CrossRef CAS.
- C. L. Qin, W. M. Cai, J. Cai, D. Y. Tang, J. S. Zhang and M. Qin, Mater. Chem. Phys., 2004, 85, 402–409 CrossRef CAS.
- B. Finnigan, D. Martin, P. Halley, R. Truss and K. Campbell, Polymer, 2004, 45, 2249–2260 CrossRef CAS.
- K. G. Gatos, J. G. M. Alcazar, G. C. Psarras, R. Thomann and J. Karger-Kocsis, Compos. Sci. Technol., 2007, 67, 157–167 CrossRef CAS.
- R. Sen, B. Zhao, D. Perea, M. E. Itkis, H. Hu, J. Love, E. Bekyarova and R. C. Haddon, Nano Lett., 2004, 4, 459–464 CrossRef CAS.
- H. Kim, Y. Miura and C. W. Macosko, Chem. Mater., 2010, 22, 3441–3450 CrossRef CAS.
- J. L. Bonczek, W. G. Harris and P. Nkedi-Kizza, Clays Clay Miner., 2002, 50, 11–17 CrossRef CAS.
- A. Y. Park, H. Kwon, A. J. Woo and S. J. Kim, Adv. Mater., 2005, 17, 106 CrossRef CAS.
- H. P. He, R. L. Frost, F. Deng, J. X. Zhu, X. Y. Wen and P. Yuan, Clays Clay Miner., 2004, 52, 350–356 CrossRef CAS.
- R. Pena-Alonso, F. Rubio, J. Rubio and J. L. Oteo, J. Mater. Sci., 2007, 42, 595–603 CrossRef CAS.
- Q. Tao, H. He, R. L. Frost, P. Yuan and J. Zhu, Appl. Surf. Sci., 2009, 255, 4334–4340 CrossRef CAS.
- K. C. Vrancken, L. Decoster, P. Vandervoort, P. J. Grobet and E. F. Vansant, J. Colloid Interface Sci., 1995, 170, 71–77 CrossRef CAS.
- J. W. Dehaan, H. M. Vandenbogaert, J. J. Ponjee and L. J. M. Vandeven, J. Colloid Interface Sci., 1986, 110, 591–600 CrossRef CAS.
- M. C. B. Salon, M. Abdelmouleh, S. Boufi, M. N. Belgacem and A. Gandini, J. Colloid Interface Sci., 2005, 289, 249–261 CrossRef CAS PubMed.
- K. Urayama, T. Miki, T. Takigawa and S. Kobjiya, Chem. Mater., 2004, 16, 173–178 CrossRef CAS.
- J. X. Song, G. Wu, J. J. Shi, Y. Ding, G. X. Chen and Q. F. Li, Macromol. Res., 2010, 18, 944–950 CrossRef CAS.
- R. Hu, V. L. Dimonie, M. S. ElAasser, R. A. Pearson, A. Hiltner, S. G. Mylonakis and L. H. Sperling, J. Polym. Sci., Part B: Polym. Phys., 1997, 35, 1501–1514 CrossRef CAS.
- K. Kojio, Y. Mitsui and M. Furukawa, Polymer, 2009, 50, 3693–3697 CrossRef CAS.
- R. W. Seymour and S. L. Cooper, Macromolecules, 1973, 6, 48–53 CrossRef CAS.
- H. S. Lee, Y. K. Wang and S. L. Hsu, Macromolecules, 1987, 20, 2089–2095 CrossRef CAS.
- Y. S. Lu, L. Tighzert, P. Dole and D. Erre, Polymer, 2005, 46, 9863–9870 CrossRef CAS.
- R. Chandra, S. P. Singh and K. Gupta, Compos. Struct., 1999, 46, 41–51 CrossRef.
- P. K. Maji, P. K. Guchhait and A. K. Bhowmick, ACS Appl. Mater. Interfaces, 2009, 1, 289–300 CAS.
- M. Kotal, S. K. Srivastava and S. K. Manu, J. Nanosci. Nanotechnol., 2010, 10, 5730–5740 CrossRef CAS PubMed.
- B. Magagula, N. Nhlapo and W. W. Focke, Polym. Degrad. Stab., 2009, 94, 947–954 CrossRef CAS.
- W. J. Choi, S. H. Kim, Y. J. Kim and S. C. Kim, Polymer, 2004, 45, 6045–6057 CrossRef CAS.
- C. Z. Liao and S. C. Tjong, J. Nanomater., 2010, 2010, 327973 Search PubMed.
- H. Acharya, M. Pramanik, S. K. Srivastava and A. K. Bhowmick, J. Appl. Polym. Sci., 2004, 93, 2429–2436 CrossRef CAS.
- C. Nyambo, P. Songtipya, E. Manias, M. M. Jimenez-Gasco and C. A. Wilkie, J. Mater. Chem., 2008, 18, 4827–4838 RSC.
- M. Kotal, T. Kuila, S. K. Srivastava and A. K. Bhowmick, J. Appl. Polym. Sci., 2009, 114, 2691–2699 CrossRef CAS.
- H. Acharya, S. K. Srivastava and A. K. Bhowmick, Compos. Sci. Technol., 2007, 67, 2807–2816 CrossRef CAS.
- M. Sabzi, S. M. Mirabedini, J. Zohuriaan-Mehr and M. Atai, Prog. Org. Coat., 2009, 65, 222–228 CrossRef CAS.
- G. Wang, S. Xu, C. Xia, D. Yan, Y. Lin and M. Wei, RSC Adv., 2015, 5, 23708–23714 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra04398d |
| ‡ The two authors contribute equal to this work. |
| This journal is © The Royal Society of Chemistry 2016 |