
Synthesis of hupehenols A, B, and E from protopanaxadiol†
Abstract
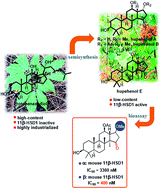
Hupehenols A–E (3–7) are bioactive octanordammarane triterpenoids, among which hupehenols B (4) and E (7) are the most potent and selective human 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1) inhibitors (IC50 = 15 and 34 nM, respectively). Herein, the hupehenols were synthesized from protopanaxadiol, the main component of Panax notoginseng. The different synthetic attempts resulted in the stereoselective synthesis of hupehenols A, B, and E for further biological exploration. Notable features of the synthesis included a regioselective epoxide-opening reaction, regioselective acetylation, and a late-stage stereoselective oxa-Michael addition. Semi-synthetic derivatization of these natural products led to the determination of their absolute configurations, a better understanding of their inherent reactivity patterns, and the required C-17 configuration for murine 11β-HSD1 inhibition. These studies provide the basis for the synthesis of 11β-HSD1 inhibitors as potential targets for the treatment of type 2 diabetes.
 Please wait while we load your content...
Please wait while we load your content...

