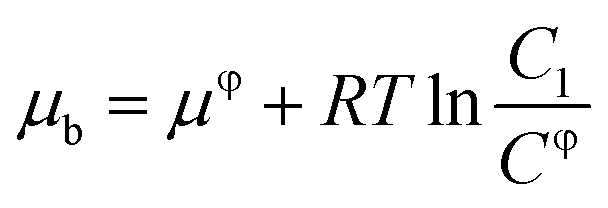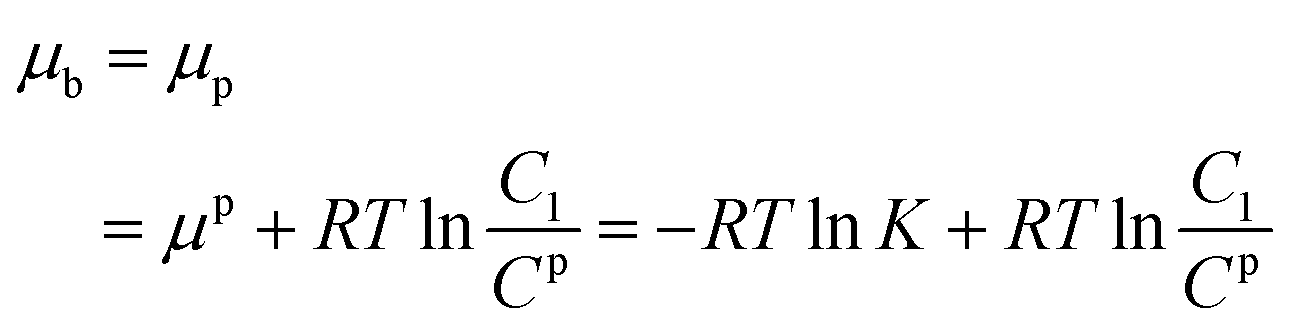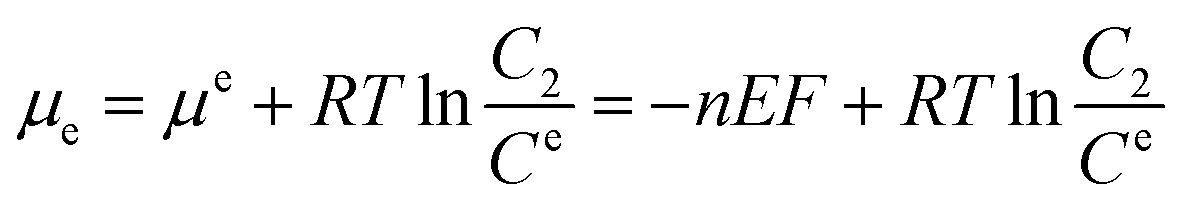DOI:
10.1039/C6RA04232E
(Paper)
RSC Adv., 2016,
6, 42869-42875
Switchable polymer reactor composed of mussel-inspired polymer that contains Au nanoparticles
Received
16th February 2016
, Accepted 11th April 2016
First published on 18th April 2016
Abstract
This study was aimed at addressing the present challenge in self-controlled catalysis, as to how to furnish smart catalysts with robust switchable ability in aqueous media. This objective was reached by developing a marine mussel-inspired polymer reactor that was capable of adapting to switch in aqueous media. This polymer reactor was composed of catalytic Au nanoparticles and a mussel-inspired polymer carrier that contained self-assembled switching interactions. The self-assembled switching interactions, by opening and closing, acted as a molecular switch for providing controlled access to the encapsulated metal nanoparticles, which caused switchable catalytic ability. In virtue of the mussel-mimicking functionality, the switchable catalytic behavior at this polymer reactor was repeatable and compatible with aqueous media, which involved neither hydrophilic/hydrophobic paradigm nor any leaching of metal nanoparticles. In this way, this polymer reactor demonstrated a robust switchable ability. This new protocol shows a promising prospect to develop robust smart catalysts for controlled catalytic processes occurring in aqueous media.
1. Introduction
Despite the pressing prospect in chemical synthesis and in inaccessible sites, self-controlled catalysis remains still a significant challenge. At the forefront of this field would be the use of smart catalysts to achieve switchable catalytic ability.1 Prominent among these are the so-called polymer reactors composed of poly(N-isopropylacrylamide) (PNIPAm)-encapsulated metal nanoparticles,2,3 which are capable of demonstrating switchable catalytic ability in water. This outcome arises from the unique thermosensitive hydrophilic/hydrophobic transition at PNIPAm, which causes either impeded or unobstructed access to the encapsulated metal nanoparticles. In this way, catalysis by polymer reactors shows a switchable process. The level of innovative thinking applied to develop functional polymer reactors over the years has been high, which may be reflected by adopting special polymers or polymeric materials containing elaborate structures as the support of metal nanoparticles.4–6 Nonetheless, the development of polymer reactors with practical potentials has been proven to be elusive, mainly because most of the reported polymers suitable for supporting metal nanoparticles are not PNIPAm, which clearly lack thermal phase transitions. Furthermore, it is difficult and even impossible to overcome the leaching of metal nanoparticles based on the repeated hydrophilic/hydrophobic switching, due to the hydration of these polymer reactors. Thus, more protocols and particularly new protocols are required.
The creation of specified structures with desired properties has been the fundamental mission in chemical and material sciences. Nature, as the best example of smart systems, has provided opportunities for scientists to seek aspirations in virtue of its concordance and functionality. A body of knowledge is already available. One of these is the self-assembled adhesive ability at marine mussel proteins,7,8 which shares a promising prospect with the struggling polymer reactors. The adhesive ability at mussels’ adhesive proteins enables mussels to strongly adhere to virtually all solid substances (even including low-fouling paraffin and Teflon). Instinctively, the strong adhesive behavior at mussels’ adhesive proteins takes place in aqueous media and these are adverse to the occurrence of adhesion. Although the exact mechanism remains to be understood, clues to the strong adhesive ability at mussels’ adhesive proteins lie in the composition of amino acids, which are rich in catechol and amino functional groups located at 3,4-dihydroxy-L-phenylalanine (dopa) and lysine segments.9 The catechol groups, when coupled with amino moieties, may dictate the adhesive proteins’ versatile interactions with substances, ranging from hydrogen bonds to covalent binding, from coordination to chelation, and from bidentate to multidentate anchoring. Dopamine-based polymers, which contain both catechol and amino groups, can form not only self-assembled architectures within the polymers but also firm attachments to almost all solids including metals. The scission of dopamine polymers would lead to complete self-healing of the polymers in virtue of the spontaneous self-assembly among dopamine and amino moieties.10 The self-healing behavior was perfectly compatible with aqueous media due to the mussel-evolved functionality. Although these reported findings are not related to catalytic applications, the occurrence of self-healing in aqueous media at mussels’ adhesive proteins provides an insight into the struggling field of self-controlled catalysis, which makes feasible switchable polymer reactors for application in aqueous media.
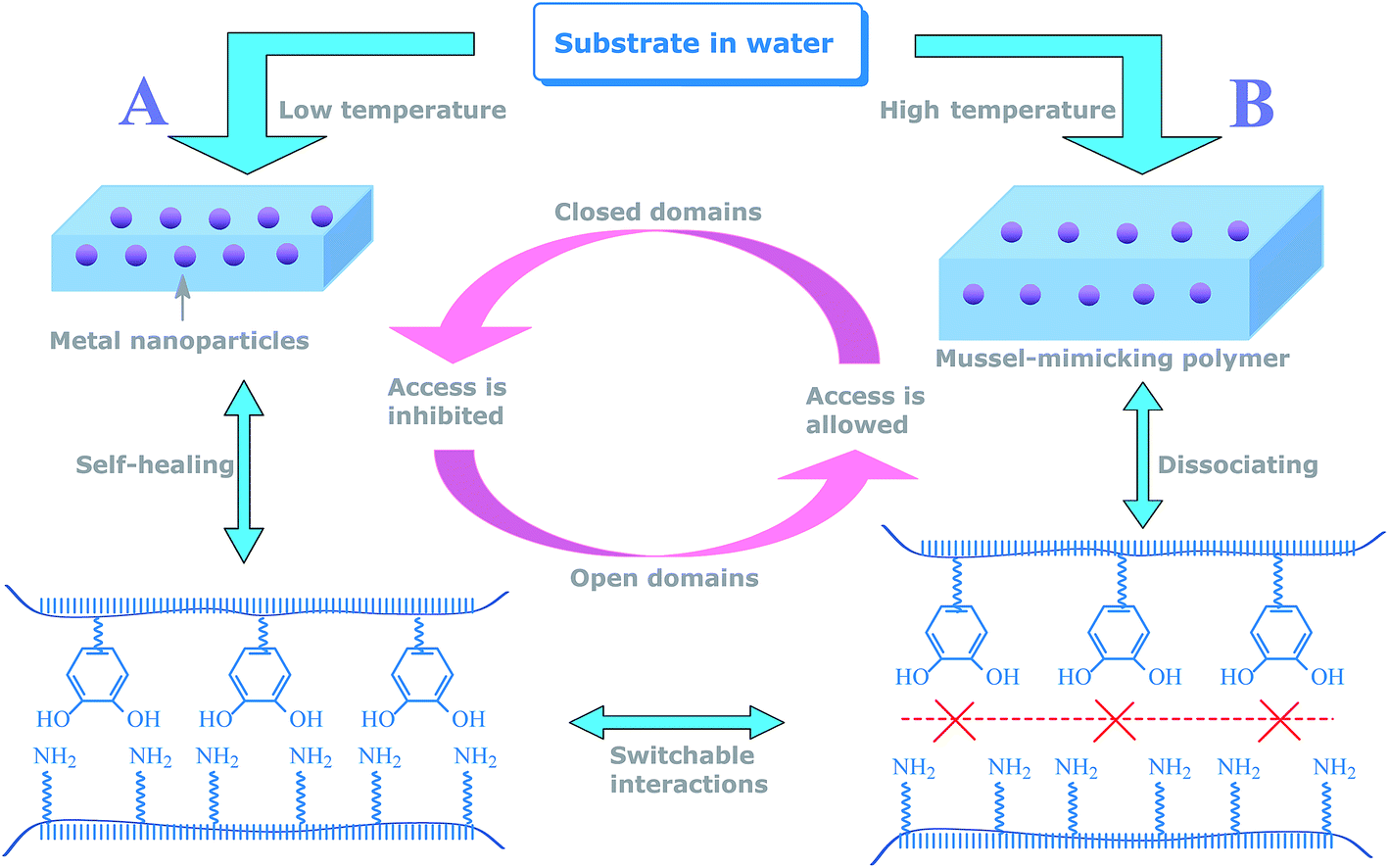
Inspired by this principle, herein, we aimed at addressing the present challenge in self-controlled catalysis by developing a mussel-inspired polymer reactor that was capable of adapting to switch in aqueous media. To the best of our knowledge, this polymer reactor represents the first report of its kind that provides switchable catalytic ability involving neither hydrophilic/hydrophobic paradigm nor any leaching of metal nanoparticles. This polymer reactor (named AuPR-S) was composed of catalytic Au nanoparticles and a mussel-inspired polymer carrier containing self-assembled switching interactions between polymeric dopamine (PDPA) and acrylamide (PAA). The switchable interaction between PDPA and PAA, by self-healing and dissociating, allowed for controlled access to the encapsulated metal nanoparticles. As proposed in Scheme 1, the closed access in this polymer reactor at relatively low temperatures would block substrates from the encapsulated metal nanoparticles, resulting in poor catalytic reactivity (Status A). Access for the substrates is, however, allowed with increasing temperature, arising from the disruption of these hydrogen bonding interactions (Status B). Given the strong anchoring between dopamine moieties and metal nanoparticles, the repeated switching in aqueous media would not cause any leaching of the encapsulated metal nanoparticles. In this way, without involving conventional hydrophilic/hydrophobic paradigm and the leaching of metal nanoparticles, this polymer reactor demonstrated robust switchable catalytic ability in aqueous media. The objective of this study is to demonstrate that polymer reactors capable of meeting the present challenge in self-controlled catalysis can be realized by using this new protocol, which suggests promising opportunities to develop robust smart catalysts for controlled catalytic processes occurring in aqueous media.
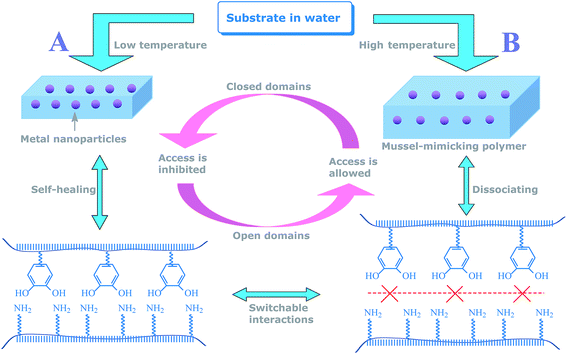 |
| | Scheme 1 Proposed mechanism for the AuPR-S reactor. | |
2. Experimental section
2.1. Preparation of polymer reactors
Unless otherwise noted, the chemicals used were of analytic grade and used as received from Sigma-Aldrich. The polymer reactor, as outlined in Scheme 1, was prepared based on the suggested design of self-healing polymers and polymer reactors.11,12 In detail, dopamine acrylamide (DPA, nomenclaturally known as N-[2-(3,4-dihydroxyphenyl)ethyl]-2-methylacrylamide, 1.0 g), acrylamide (AA, 1.61 g), chloroauric acid (1.86 g), divinylbenzene (0.11 g) and AIBN (0.22 g) were dissolved in dimethylformamide (20.0 mL). After being dispersed and deoxygenated with sonication and argon, this mixture system was heated up to 60 °C and allowed to react for 10 h with stirring. The resulting viscous system was treated with methanol and diethyl ether profusely, resulting in the formation of solid reactor precursors. The encapsulated ionic Au was then reduced by an excess of sodium borohydride (tenfold, with regard to the molar amount of ionic Au). The resulting polymer reactor was washed with methanol and water, dried in flowing argon, and then ground into a size of ca. 60-mesh. In this way, the polymer reactor was prepared (i.e. AuPR-S).
For a contrastive purpose, two controls, named AuPR-N, and PR-S, were also prepared under comparable conditions. AuPR-N was the non-responsive polymer reactor prepared as AuPR-S but without using DPA (herein, the suffix N means non-responsive properties in contrast to the suffix S, for switchable characteristics in AuPR-S). PR-S was the polymeric carrier of AuPR-S and prepared without using Au. For convenient discussion, all of the prepared polymer reactors and carrier are mentioned afterward as the conceptual reactors.
2.2. Characterization
The TEM images of the above-prepared reactors were obtained using transmission electron microscopy (TEM) (JEM-2100, Japan). Samples dispersed in alcohol were placed onto a 200-mesh copper TEM grid (3 mm) and the acceleration voltage used was 200 kV. The infrared spectra were taken in KBr-pressed tablets using FTIR apparatus (Nicolet MX-1E, USA). The absorption bands of surface plasma resonance (SPR) were recorded using a Lambda 25 UV spectrometer (USA). The Au content in these reactors was detected using energy-dispersive spectroscopy (EDS) apparatus (MIRA3-XMU, USA).
2.3. Switchable interactions
The switchable interaction between PDPA and PAA was studied as a function of temperature using dynamic light scattering (DLS) (ZS-ZEN3600, UK).13 For equilibrium, all the samples concerned were kept at the specified temperatures for at least 10 min before acquiring hydrodynamic radii (Rh). By a comparison between the switchable AuPR-S and PR-S and the non-responsive AuPR-N, the contribution of the switchable interactions was therefore reflected by the relative change of the swelling ratio (Rc):13,14
Herein, Rd is the particle size of dried particles, the subscript S means the switchable reactors and N represents the non-responsive reactor. The term Rh − Rd/Rd presents the swelling ratio of a specified reactor.
2.4. Catalytic testing
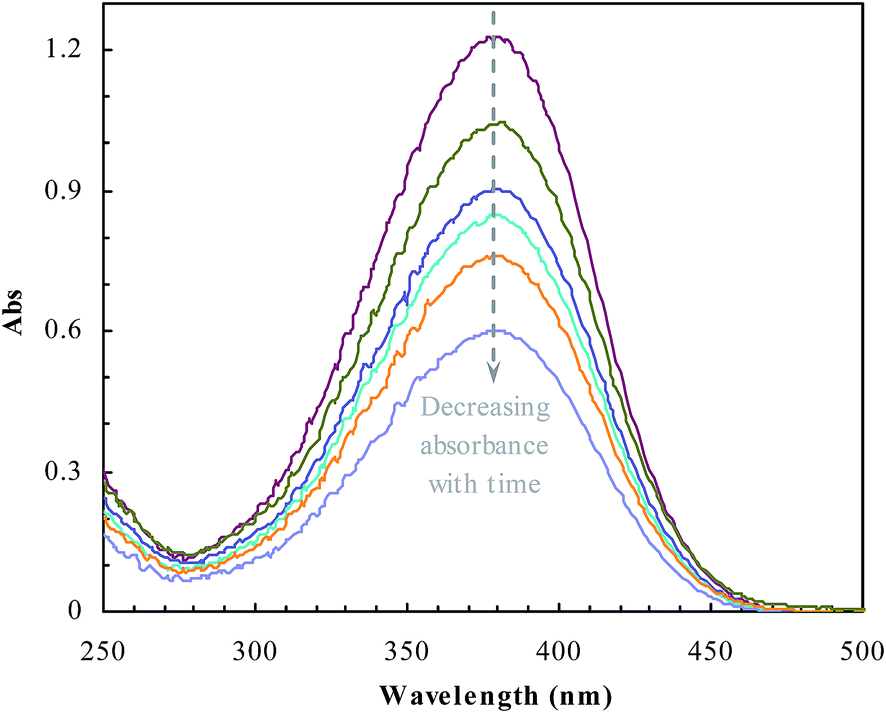
The catalytic properties of the prepared reactors were evaluated using the common model reaction of reducing p-nitroaniline with sodium borohydride.15 p-Nitroaniline was added into NaBH4 aqueous solution with an initial concentration of 0.1 μmoL mL−1 (total volume of 3 mL) (NaBH4, tenfold with regard to the molar amount of p-nitroaniline). The amount of polymer reactors used in every test was 0.8 mg mL−1. The reduction of p-nitroaniline was monitored spectrophotometrically (380 nm, cf. Fig. 1). The catalytic activities of these polymer reactors were achieved from the mass balance of p-nitroaniline based on the average of triple runs.
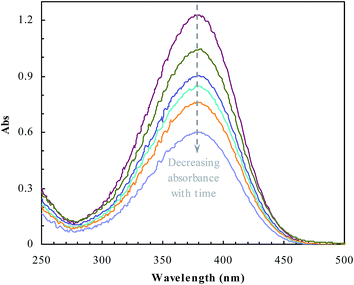 |
| | Fig. 1 UV-vis spectra changing with the reduction of p-nitroaniline by sodium borohydride. | |
2.5. Electrochemical testing
Electrochemical testing was further performed to achieve information on the interaction between the prepared reactors and substrate.16 Using an electrochemical workstation equipped with a conventional three-electrode configuration (Au plate working electrode, Pt wire counter electrode, and Ag/AgCl reference electrode) (CHI 760E, China), polymer reactors (10.0 mg) that pre-absorbed ca. 1 μg p-nitroaniline were placed into a cuvette encircled by a diffusion-eliminating sonication apparatus (supporting electrolyte of 0.01 mmol mL−1 KCl, 10 mL). The desorption behavior of the absorbed substrate was monitored by consecutively scanning with cyclic voltammetry until a stable desorption/reduction profile was reached (scanning range −0.4 to −1.0 V, rate 1 mV s−1).
3. Results and discussion
3.1. Characterization
The polymer reactor AuPR-S, as aforementioned, was composed of Au nanoparticles and a mussel-inspired polymer carrier composite of PDPA and PAA. The FTIR spectrum was first used to identify the polymeric composition, as shown in Fig. 2. Three major bands (i.e. 3000–3650, ∼1750 and 800–1400 cm−1) appeared in the FTIR spectrum, corresponding to the stretching of O–H/N–H, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–N/C–C, respectively.17 Given the constituent PDPA and PAA, which both contain O–H/N–H, CO and C–N/C–C bonds, it is therefore difficult to directly identify one specified component from AuPR-S. As such, we also included the FTIR spectra of the two controls described above (i.e. PR-S and AuPR-N, with carriers made of PDPA–PAA, and PAA, respectively), along with that of PDPA, in Fig. 2. AuPR-S exhibited almost the same spectrum as that of PR-S and contained the major bands of both AuPR-N and the PDPA polymer. In conjunction with the preparation process (cf. Section 2.1), this outcome indicates that AuPR-S was the polymer reactor composite of PDPA and PAA.
O and C–N/C–C, respectively.17 Given the constituent PDPA and PAA, which both contain O–H/N–H, CO and C–N/C–C bonds, it is therefore difficult to directly identify one specified component from AuPR-S. As such, we also included the FTIR spectra of the two controls described above (i.e. PR-S and AuPR-N, with carriers made of PDPA–PAA, and PAA, respectively), along with that of PDPA, in Fig. 2. AuPR-S exhibited almost the same spectrum as that of PR-S and contained the major bands of both AuPR-N and the PDPA polymer. In conjunction with the preparation process (cf. Section 2.1), this outcome indicates that AuPR-S was the polymer reactor composite of PDPA and PAA.
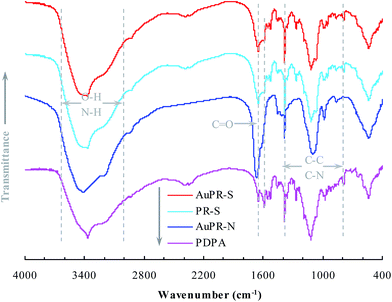 |
| | Fig. 2 FTIR spectra of the prepared polymer reactors (for polymer reactors, Au means Au nanoparticles, PR suggests polymer reactors, S stands for switchable properties, and N represents non-responsive properties). | |
SPR and EDS analyses were further carried out to detect the presence of Au nanoparticles in the prepared reactors, as shown in Fig. 3. Compared with the Au-lacking reactor PR-S, the Au-containing reactors AuPR-S and AuPR-N exhibited typical SPR bands of Au nanoparticles at ca. 510 nm (Fig. 3a).18,19 The Au content burdened in AuPR-S and AuPR-N was ca. 3.9 wt% (Fig. 3b). Fig. 4 presents the TEM images exhibiting the morphology of Au nanoparticles encapsulated in these prepared reactors. Au nanoparticles with a size of ca. 10 nm were encapsulated in the polymeric carriers. Hence, these polymer reactors were prepared in the desired form.
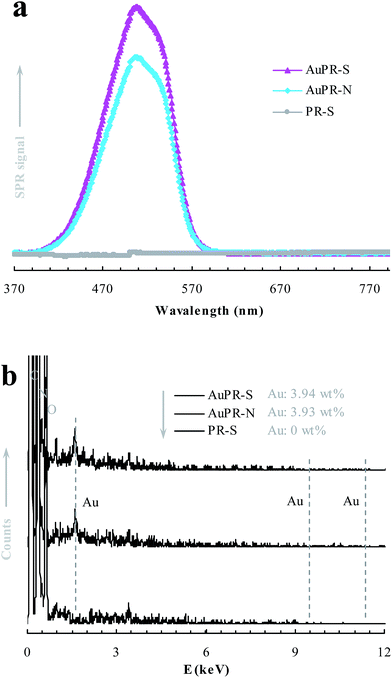 |
| | Fig. 3 SPR (a) and EDS (b) spectra of the prepared polymer reactors. | |
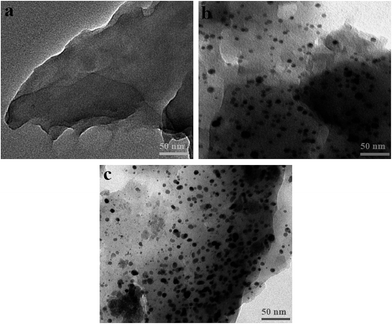 |
| | Fig. 4 TEM images of the metal nanoparticles contained in the prepared polymer reactors ((a) PR-S, (b) AuPR-S, and (c) AuPR-N). | |
3.2. Switchable interactions
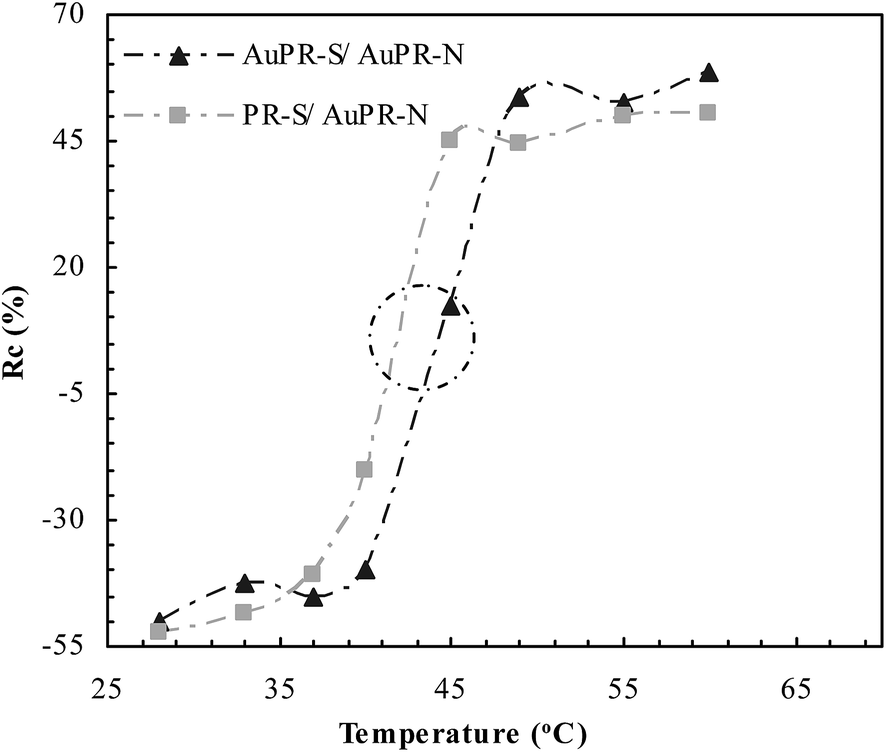
The switchable interaction between PDPA and PAA was studied as a function of temperature, as shown in Fig. 5. Compared with the non-responsive AuPR-N, the change (Rc) of the swelling ratio at the switchable AuPR-S and PR-S showed a significant dependence on temperature. The dramatic change of Rc at AuPR-S and PR-S appeared at ca. 43 °C (marked with a circle). Below this temperature, AuPR-S and PR-S showed a low Rc value associated with the complementary interaction between PDPA and PAA, which inhibited the swelling of the polymers. On the contrary, above that temperature, AuPR-S and PR-S showed a significantly increased Rc value in response to the dissociation of the PDPA–PAA interaction. This outcome therefore indicates that the switchable interaction between PDPA and PAA has been engineered to the prepared AuPR-S. In conjunction with the development of this polymer reactor (cf. Scheme 1), the switchable interaction in AuPR-S potentially makes feasible switchable catalytic ability.
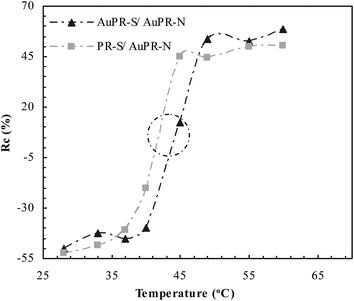 |
| | Fig. 5 DLS curves of the prepared polymer reactors. | |
3.3. Catalysis and robust switchable ability
The catalytic properties of these polymer reactors are presented in Fig. 6. PR-S did not show significant catalysis because of the lack of catalytic Au nanoparticles. In contrast, AuPR-S and AuPR-N showed significant catalytic reactivity where the conversion rapidly increased with time. For verifying the switchable catalytic ability, two representative temperatures, 30 and 50 °C (either lower or higher than the transition temperature of AuPR-S (i.e. 43 °C, cf. Fig. 3)), were selected for a contrastive study. The purpose of selecting such temperatures was to ensure that the switchable statuses A and B can be covered (cf. Scheme 1). As shown in Fig. 6, the catalytic activities at the non-responsive AuPR-N increased with the temperature. The catalytic activities at AuPR-S were, however, lower than that at AuPR-N at 30 °C and then became higher than that of AuPR-N at 50 °C. AuPR-S demonstrated switchable catalytic ability, as expected.
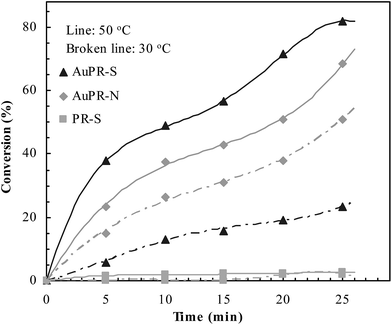 |
| | Fig. 6 Catalytic activities of the prepared polymer reactors. | |
The ability to repeat switching at AuPR-S is presented in Fig. 7. The catalysis can be made to repeat switching between the two statuses A and B without substantially decreasing the catalytic activities (cf. Scheme 1). Owing to the strong anchoring between dopamine moieties and metal nanoparticles, no leaching of metal nanoparticles had been detected during the repeated switching. AuPR-S demonstrated the robust switchable ability in aqueous solution, as expected.
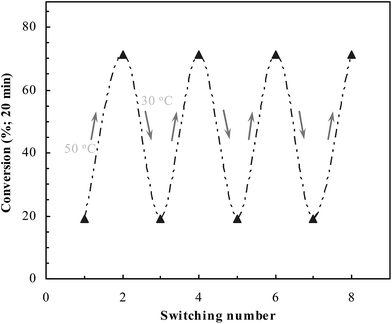 |
| | Fig. 7 Repeating of the switchable catalytic behavior at AuPR-S. | |
3.4. Dynamic interactions and switchable access
Electrochemical testing was further carried out to interrogate the dynamic interaction between the prepared reactors and substrate. It is known that the potential to reduce or oxidize a binding molecule depends on the binding constant (between the reactors and the substrate). A relatively stronger binding state will need more energy to overcome the binding, thereby resulting in a larger redox potential. As schematically presented in Scheme 2, the substrate molecules (B) in the system would normally involve desorption, diffusion to the surface of the electrodes, and electrochemical reaction. In the event that the diffusion is eliminated with sonication, the electrochemical reaction of the substrate would be closely associated with the desorption behavior. Based on thermodynamic theories, the chemical potential for the substrate in bulk solution would be:| |
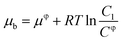 | (1) |
where μφ and Cφ are the standard chemical potential of the substrate and the corresponding standard concentration, and C1 is the practical concentration. R is the gas constant (8.314 J mol−1 K−1) and T is the temperature. Correlating eqn (1) with the absorption/desorption equilibrium of the substrate would give eqn (2):| |
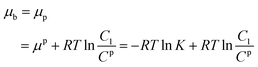 | (2) |
where the superscript or subscript p represents the polymer reactors, implicating the dynamic interaction between the reactors and the substrate. K is the equilibrium constant of absorbing the substrate onto the polymer reactors, suggesting the affinity of the polymer reactors for the substrate. Like the mathematical treatment in bulk solution, the use of thermodynamic theories for the substrate on the surface of the working electrode would give eqn (3):| |
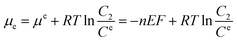 | (3) |
where E is the redox potential of the substrate, n is the molar number of the electrons transferred during the redox process, and F is the Faraday constant (96![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 C mol−1). Ce is the standard concentration of the substrate on the surface of the working electrode, and C2 is the corresponding practical concentration on the surface. The deduction of eqn (2) with (3) leads to eqn (4):
485 C mol−1). Ce is the standard concentration of the substrate on the surface of the working electrode, and C2 is the corresponding practical concentration on the surface. The deduction of eqn (2) with (3) leads to eqn (4):| |
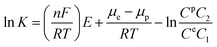 | (4) |
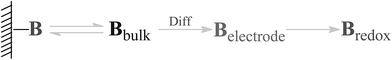 |
| | Scheme 2 Schematic presentation of an electrochemical process with a binding molecule B. | |
After eliminating the concentration gradient of the substrate in bulk solution with sonication, consecutive scanning with cyclic voltammetry in a fixed and low rate up to a stable desorption/redox profile would give eqn (5):
| |
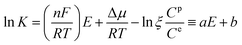 | (5) |
where
ζ represents the ratio of
C2/
C1 at the desorption/redox equilibrium of the substrate, and
a and
b are constants that include invariable items. The interaction of the substrate with different polymer reactors under comparable conditions leads to the expression:
| | |
ΔlnK = aΔE
| (6) |
It is therefore clear that the binding state between the polymer reactors and the substrate is closely associated with the redox potential of the binding substrate. A relatively stronger binding state between the polymer reactors and the substrate may be reflected by a larger redox potential of the binding substrate. As such, the electrochemical testing was carried out in accordance with the paradigm, as shown in Fig. 8. Given the responsive properties at AuPR-S, we further selected 30 and 50 °C for a contrastive study. p-Nitroaniline that was attached to AuPR-S at 30 °C exhibited a desorption/reduction peak at −865 mV. In contrast, this peak at 50 °C shifted to a smaller position (−817 mV). AuPR-S showed a stronger interaction with p-nitroaniline at 30 °C than at 50 °C. The interaction offered by AuPR-S showed adjustable properties.
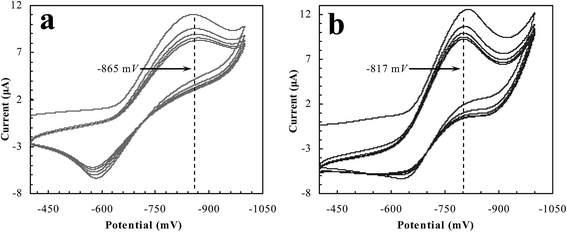 |
| | Fig. 8 Reduction profiles with substrate desorbing from AuPR-S ((a) 30 °C and (b) 50 °C). | |
For further addressing the interaction, Table 1 provides the desorption/reduction potentials with p-nitroaniline desorbing from all of the prepared polymer reactors. Despite the encapsulated metal nanoparticles, AuPR-S exhibited almost the same desorption/reduction potentials as those of PR-S. The potentials at AuPR-S and PR-S exhibited significant shifts with increasing temperature. In contrast, the non-responsive AuPR-N did not show a significant change at the desorption/reduction potential, regardless of the increasing temperature. This outcome strongly suggests that the switchable catalytic ability at AuPR-S lied in the responsive polymeric carrier, which allowed for switchable access to the encapsulated metal nanoparticles. The properties, when coupled with the mussel-mimicking functionality, therefore dictated the AuPR-S reactor robust switchable ability.
Table 1 Reduction potentials with substrate desorbing from all the prepared polymer reactors (mV)
| Polymer reactor |
30 °C |
50 °C |
Delta |
| AuPR-S |
−865 |
−817 |
48 |
| PR-S |
−861 |
−812 |
49 |
| AuPR-N |
−836 |
−831 |
5 |
4. Conclusions
In this study, we aimed at addressing the present challenge in self-controlled catalysis by reporting a marine mussel-inspired polymer reactor that was capable of adapting to switch in aqueous media. This polymer reactor was composed of catalytic Au nanoparticles and a mussel-inspired carrier containing self-assembled switching interactions that allowed for controlled access to the encapsulated metal nanoparticles. At relatively low temperatures, this polymer reactor showed poor catalytic activities due to the closed access, which blocked substrates from the catalytic metal nanoparticles. In contrast, this polymer reactor showed significant catalytic activities at relatively high temperatures, in response to the opening at the access. In virtue of the mussel-mimicking functionality, the repeated switching did not cause the leaching of the encapsulated metal nanoparticles. There was not any substantial decrease in the catalytic activities after repeated switching, either. Unlike known polymer reactors or smart catalysts, the switchable catalytic behavior at this polymer reactor was compatible with aqueous media, which did not involve any hydrophilic/hydrophobic paradigm. It is therefore confirmed that polymer reactors having robust switchable ability can be prepared by using this biomimetic protocol. Future development in this field will significantly increase the applications and lead to the appearance of novel smart catalysts and catalytic materials.
Acknowledgements
The authors want to express their gratitude to the National Science Foundation of China (No. 51473070, 21403091, and 51402128). Thanks also should be expressed to Jiangsu Province for the support under the innovation/entrepreneurship program (Surencaiban [2015]26) and the scientific research programs (BK20130486 and SBK2014041874).
References
- S. Cao, J. Chen, Y. Ge, L. Fang, Y. Zhang and A. P. F. Turner, Chem. Commun., 2014, 50, 118–120 RSC.
- G. Marcelo, M. Lopez-Gonzalez, F. Mendicuti, M. P. Tarazona and M. Valiente, Macromolecules, 2014, 47, 6028–6036 CrossRef CAS.
- J. Zhang, M. Zhang, K. Tang, F. Verpoort and T. Sun, Small, 2014, 10, 32–46 CrossRef CAS PubMed.
- M. Zhao, K. Deng, L. He, Y. Liu, G. Li, H. Zhao and Z. Tang, J. Am. Chem. Soc., 2014, 136, 1738–1741 CrossRef CAS PubMed.
- G. Yun, Z. Hassan, J. Lee, J. Kim, N.-S. Lee, N. H. Kim, K. Baek, I. Hwang, C. G. Park and K. Kim, Angew. Chem., Int. Ed., 2014, 53, 6414–6418 CrossRef CAS PubMed.
- P. Xu, X. Han, B. Zhang, Y. Du and H.-L. Wang, Chem. Soc. Rev., 2014, 43, 1349–1360 RSC.
- B. P. Lee and S. Konst, Adv. Mater., 2014, 26, 3415–3419 CrossRef CAS PubMed.
- G. P. Maier, M. V. Rapp, J. H. Waite, J. N. Israelachvili and A. Butler, Science, 2015, 349, 628–632 CrossRef CAS PubMed.
- H. Lee, B. P. Lee and P. B. Messersmith, Nature, 2007, 448, 338–341 CrossRef CAS PubMed.
- B. K. Ahn, D. W. Lee, J. N. Israelachvili and J. H. Waite, Nat. Mater., 2014, 13, 867–872 CrossRef CAS PubMed.
- F. Luo, T. L. Sun, T. Nakajima, T. Kurokawa, Y. Zhao, K. Sato, A. B. Ihsan, X. Li, H. Guo and J. P. Gong, Adv. Mater., 2015, 27, 2722–2727 CrossRef CAS PubMed.
- Q. M. Kainz and O. Reiser, Acc. Chem. Res., 2014, 47, 667–677 CrossRef CAS PubMed.
- R. Luo, M. Zhu, X. Shen and S. Li, J. Catal., 2015, 331, 49–56 CrossRef CAS.
- Y. Han, X. Yuan, M. Zhu, S. Li, M. Whitcombe and S. A. Piletsky, Adv. Funct. Mater., 2014, 24, 4996–5001 CrossRef CAS.
- T. Ji, L. Chen, L. Mu, R. Yuan, M. Knoblauch, F. S. Bao and J. Zhu, Appl. Catal., B, 2016, 182, 306–315 CrossRef CAS.
- Q. Li, M. Zhu, X. Shen and S. Li, RSC Adv., 2015, 5, 34985–34991 RSC.
- Y. Zhou, M. Zhu and S. Li, J. Mater. Chem. A, 2014, 2, 6834–6839 CAS.
- D. S. Sheny, J. Mathew and D. Philipb, Spectrochim. Acta, Part A, 2011, 79, 254–262 CrossRef CAS PubMed.
- Y. Hao, X. Shao, B. Li, L. Hu and T. Wang, Mater. Sci. Semicond. Process., 2015, 40, 621–630 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.