
Trehalose-8-hydroxyquinoline conjugates as antioxidant modulators of Aβ aggregation†
Valentina Oliveriab,
Francesco Belliac,
Giuseppa Ida Grassoc,
Adriana Pietropaolod and
Graziella Vecchio*a
aDipartimento di Scienze Chimiche, Università di Catania, Viale A. Doria 6, 95125, Catania, Italy. E-mail: gr.vecchio@unict.it
bConsorzio Interuniversitario di Ricerca in Chimica dei Metalli nei Sistemi Biologici C.I.R.C.M.S.B, Unità di Ricerca di Catania, 95125 Catania, Italy
cIstituto di Biostrutture e Bioimmagini, CNR, Via P. Gaifami 18, 95126 Catania, Italy
dDipartimento di Scienze della Salute, Università di Catanzaro, Viale Europa, 88100 Catanzaro, Italy
First published on 5th May 2016
Abstract
Trehalose has been proven to provide protection to different proteins to various extents, inhibiting at elevated concentrations the aggregation of proteins involved in neurodegenerative disorders such Alzheimer's, Parkinson's and Huntington's diseases. Moreover, 8-hydroxyquinolines have also been found to protect against neurodegeneration in animal models. Here, we evaluated trehalose-8-hydroxyquinoline conjugates as antioxidants and inhibitors of self-induced Aβ aggregation. All trehalose derivatives demonstrate a significant in vitro antioxidant capacity and antiaggregant ability. The conjugation of trehalose with 8-hydroxyquinoline induces synergistic effects that lead to superior antiaggregant properties. In particular, 6,6′-difunctionalized trehalose is more effective than the corresponding 6-monofunctionalized compound suggesting that grafting two 8-hydroxyquinoline moieties on the disaccharide scaffold produces a better-performing antiaggregant compound. In silico data shed light on the binding modes of the 6-monofunctionalized and 6,6′-difunctionalized trehalose with Aβ. In particular, different to the 6-monofunctionalized compound, which mostly induces ring–ring or salt-bridge interactions also involving the glucose ring of trehalose, the 6,6′-difunctionalized derivative induces pi-stacking interactions involving 8-hydroxyquinoline moieties and the aromatic rings of F4, H14 and F20 of Aβ. The obtained results are encouraging and highlight the potential of trehalose derivatives as therapeutics for amyloid-related pathologies.
Introduction
Neurodegenerative diseases, such as Alzheimer's (AD), Parkinson's (PD), Huntington's (HD), and Wilson's (WD) diseases, are multifactorial in their etiopathology. Although the pathogenesis of AD is not fully understood, many factors such as the formation of β-amyloid deposits, τ-protein, oxidative stress and dyshomeostasis of biometals, are considered to play pivotal roles.1–3For this reason, multitarget-directed molecules have recently been proposed as agents more adequate for addressing the complexity of AD.4 Preventing or reducing Aβ aggregation is one of therapeutic strategies under development or in clinical trials for the treatment of AD. Preventing Aβ aggregation can be accomplished using a variety of inhibitors such as small organic molecules, including curcumin,5 8-hydroxyquinolines (OHQs),6 but also carbohydrates such as cyclodextrins7 (CyD) and trehalose (Tre) have been shown to stabilize protein folding and inhibit protein aggregation.
Tre (α-D-glucopyranosyl-(1,1)-α-D-glucopyranoside or trehalose) is a non-reducing disaccharide that generally exists in yeast, bacteria, fungi and invertebrates. This disaccharide is known to impart stability to organisms tolerating environmental stress such as anhydrobiosis and cryobiosis by protecting proteins and cells.8 Recent studies suggest that Tre can attenuate the insulin amyloid formation in vitro9 and aggregation of Aβ and tau, associated with AD pathology.10–12 Furthermore, Tre is also effective in inhibiting polyglutamine mediated protein aggregation and fibrillation of α-synuclein and alleviating the symptoms in models of HD13 and PD.14 Because of Tre is a relevant biological molecule in several species, considerable efforts have been invested in the design of Tre analogs, mimetics and derivatives as potential fungicides and antibiotics (especially for M. Tubercolosis).15 In some cases, 6,6′-difunctionalized derivatives are biologically more active than mono-substituted compounds and show interesting biological effects.16 To quote an example, a 6,6′-difunctionalized derivative known as Brartemicin, recently isolated from the actinomycete of the genus Nonomuraea inhibits cancer cell invasion.17,18
This context has inspired us to synthesize and analyze a new difunctionalized trehalose-8-hydroxyquinoline conjugate in depth. 8-Hydroxyquinolines have the capability to directly influence the self-association and in vivo toxicity of Aβ.19 These properties are complementary to the other mechanisms of action, described for two known OHQ derivatives (clioquinol and PBT2), associated with a ionophoric activity resulting in promotion of intracellular metal uptake by neurons.20
We have previously functionalized OHQs with sugars to ameliorate their properties. These glycoconjugates have interesting features including the enhancement of solubility, reduced toxicity, and multifunctionality.21–24 In particular, we have studied the metal-binding properties and the ability to inhibit metal-induced aggregation of Aβ and β-lactoglobulin A.25,26
Herein, we report the synthesis and characterization of 6,6′-dideoxy-6,6′-di[[(8-hydroxyquinoline)-2-carboxyl]amino]-α,α′-trehalose (Tre(HQ)2, Fig. 1). Moreover, we tested the ability of the new compound Tre(HQ)2, the analogous monosubstituted 6-deoxy-6-[[(8-hydroxyquinoline)-2-carboxyl]-amino]-α,α′-trehalose26 (TreHQ, Fig. 1) and 6-deoxy-6-[[(8-hydroxyquinoline)-2-methyl-amino]-α,α′-trehalose26 (TreRHQ, Fig. 1) to scavenge free radical species and inhibit Aβ aggregation. We also tested a bis(8-hydroxyquinoline) compound (HQ)2, 8-hydroxyquinolinecarboxamide (HQ) and Tre to examine the role of the sugar and/or quinoline moieties in inhibiting free radical species and protein aggregation.
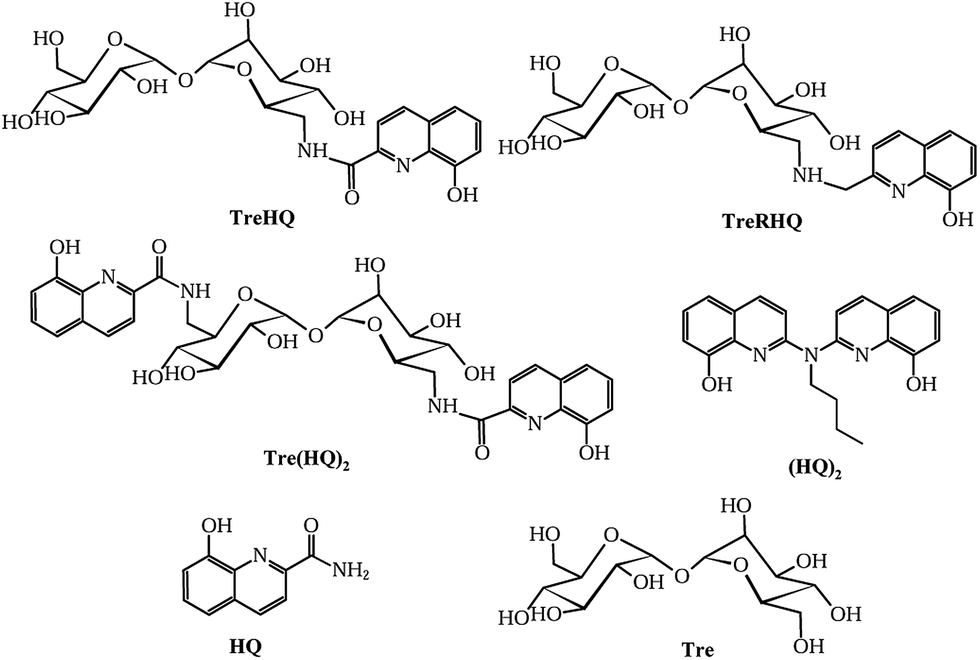 | ||
| Fig. 1 Chemical structure of the investigated compounds: trehalose-8-hydroxyquinoline conjugates and their parent compounds (HQ and Tre). | ||
The activity of the Tre-HQ derivatives toward Aβ, and radical species confirms that the conjugation of Tre with one or more HQ moieties represents a promising strategy to design new molecules that target and modulate pathological features of neurodegenerative disorders.
These findings may contribute to the understanding of the features that interfere with protein aggregation and, thus, these studies might address those seeking better inhibitors.
Results and discussion
Synthesis and characterization
6,6′-Diamino-6,6′-dideoxytrehalose was obtained from selective bromination of Tre at C-6,6′, followed by azide displacement, and reduction (6,6′-dibromo → 6,6′-diazido → 6,6′-diamino). This latter compound was coupled with two equivalents of 8-hydroxyquinoline-2-carboxylic acid in the presence of DCC and HOBt to give the difunctionalized product in good yield. ESI-MS and NMR confirmed the identity of the product.ESI-MS experiments provided evidence that the compound was difunctionalized because the spectra display the peaks resulting from the singly charged ions at m/z = 683.1 and 705.2 (assigned to [M + H]+ and [M + Na]+).
1H NMR spectrum of Tre(HQ)2 displays the signals due to the quinoline ring and Tre moiety, the former resonating in the aromatic region between 8.38 and 7.14 ppm (Fig. 2). 2D spectra allowed unambiguous assignment of all protons and carbons (Fig. S1 and S2†). Because of the symmetry of the molecule, the protons of the glucose rings as well as those of quinoline moieties are equivalent. The Hs-1 of Tre resonate at 5.08 ppm, Hs-6 and Hs-5 are shifted downfield at 3.75 and 4.08 ppm upon functionalization. As observed in the 13C NMR spectrum (Fig. S3†), C-1 carbons resonate at δ = 95.7 ppm and it is noteworthy that C-6 carbons are significantly shifted at 41.1 ppm owing to the derivatization. The UV-vis spectrum of Tre(HQ)2 in MOPS buffer (pH 7.4) is characterized by absorption bands at 253, 307, and 351 nm (Fig. S4†). The UV bands of this compound are assigned to the π–π* and n–π* transitions, similarly to other alike systems.27
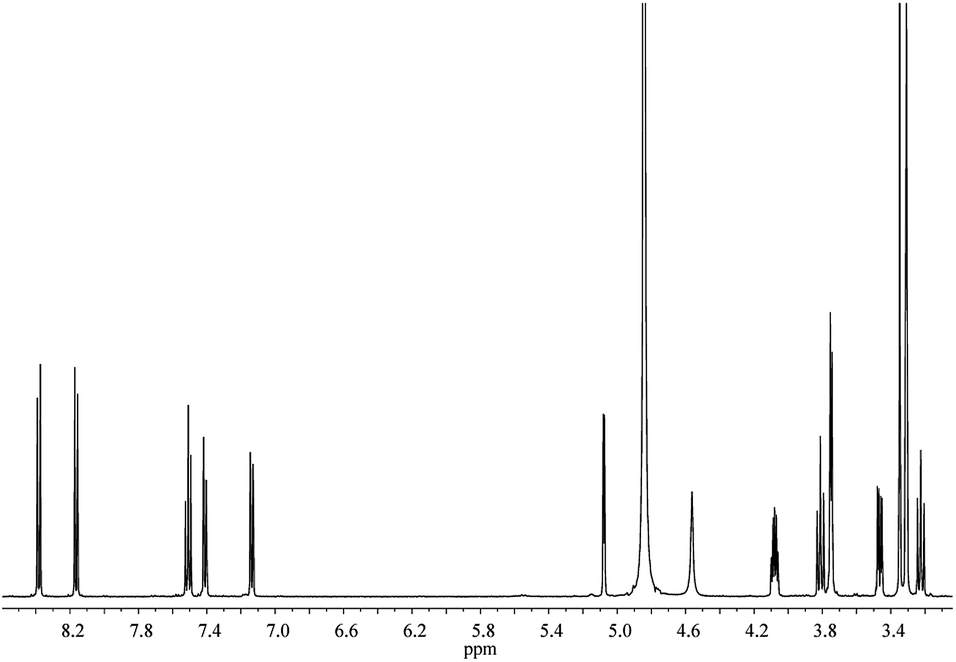 | ||
| Fig. 2 1 H NMR spectrum of Tre(HQ)2 in CD3OD at 500 MHz. | ||
Antioxidant effect
Among the multiple factors involved in AD, oxidative stress appears to be a primary progenitor of AD pathogenesis and progression.28 Recent researches have demonstrated that oxidative damage is an event that precedes the appearance of other pathological hallmarks (i.e. plaques).29 Thus, drugs that scavenge free radicals could be particularly effective in the treatment of AD and several antioxidants have been tested in clinical trials. Nevertheless, antioxidant cocktails or multifunctional antioxidants combined with metal chelators or other drugs may have successful synergistic effects.30The ability of Tre(HQ)2 to scavenge free radicals was determined by ABTS radical assay. Trolox, a water soluble vitamin E analogue, was used as a standard, and the results are expressed as Trolox Equivalent Antioxidant Capacity (TEAC). The antioxidant efficiency of the phenolic compounds is related to the number of hydroxyl groups in the molecule and also to other effects such as hydrogen atom donating ability of the compounds. The nature of substituent effects (electron-withdrawing, electron donator, inductive effects) has an influence on the H-donating ability of hydroxyl group. As a consequence, the functionalization of HQ moiety to append a sugar could influence the scavenging ability, increasing or reducing TEAC values as observed in the case of other derivatives.22 The TEAC values of Tre(HQ)2 are reported in Fig. 3. To obtain a more complete picture of the role of the HQ moiety and, in particular, of the phenolic group, TEAC values of the parent compounds HQ and (HQ)2 were also determined as well as those of TreHQ and TreRHQ were reported (Fig. 3). The results showed that phenolic groups play an important role in the antioxidant activity of the tested compounds. It is noteworthy that phenolic compounds exhibit a wide range of biological functions that are attributed to their radical-scavenging activity.
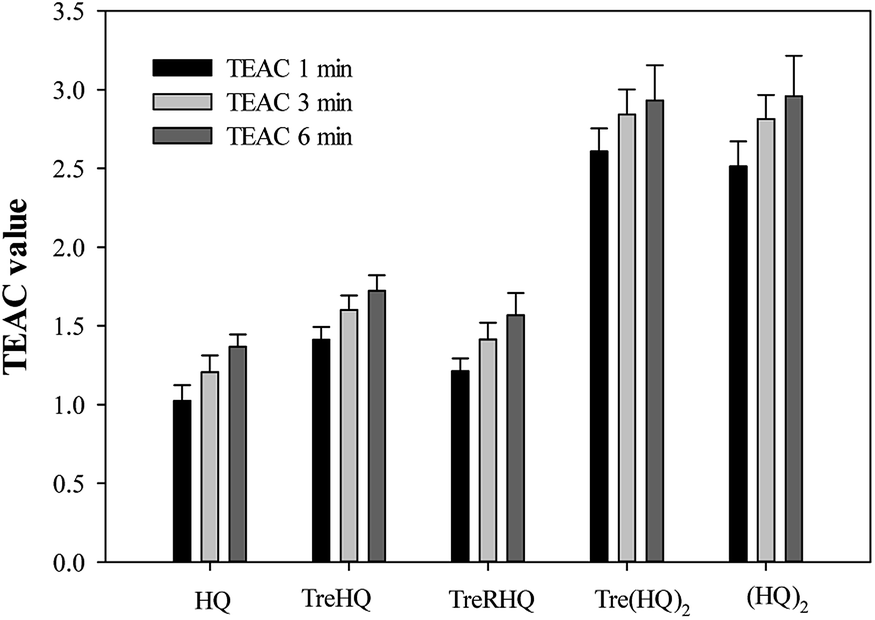 | ||
| Fig. 3 TEAC values at 1, 3, and 6 min for Tre derivatives, HQ and (HQ)2. Means are the average of three independent trials, and error bars show standard deviations. | ||
All compounds are more active than Trolox and this higher activity is generally related to a good protective function against oxidative stress. Furthermore, the difunctionalized systems have better antioxidant activity than compounds with only one HQ moiety. Among all of the derivatives tested, Tre(HQ)2 showed the best antioxidant activity, which was close to that of (HQ)2. At the end point (after 6 min), Tre(HQ)2 showed a TEAC value of 2.9, which is higher than the reported value for gallic acid (2.6), a naturally occurring polyphenol antioxidant, which has recently been shown to have potential health effects. These data suggest that Tre(HQ)2 could be a powerful antioxidant with activity comparable to that of several polyphenols.
Antiaggregant effect
Preventing or reducing Aβ aggregation is being actively pursued as a therapeutic strategy under development or in clinical trials for the treatment of AD. Inhibiting Aβ aggregation might suppress Aβ-induced neurotoxicity. To this end, the intervention of TreHQ, TreRHQ and Tre(HQ)2 on the aggregation of Aβ1–42 was explored by testing different Aβ/compound ratios ranging from 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 1:10. When the aggregation process was monitored by a turbidimetric assay (Fig. 4), TreRHQ and Tre(HQ)2 were able to significantly reduce the aggregation extent of an equimolar amount of Aβ1–42. At higher concentrantions of the tested compounds, all the Tre-HQ glycoconjugates were equally proficient in hampering the formation of both amorphous and fibrillar Aβ aggregates (Fig. 4). This ‘levelling effect’ in the turbidimetric assay is not observed when the formation amyloid-type fibrous aggregates was evaluated by means of the Thioflavin T (ThT) fluorimetric assay. ThT is an organic dye whose fluorescence intensity greatly enhances upon binding to amyloid fibrils both (Fig. S5†) in vitro and ex vivo. For this reason, the aggregation of Aβ1–42 in vitro has been monitored by measuring the fluorescence emission of ThT co-incubated with the amyloid peptide. TreHQ, TreRHQ and Tre(HQ)2, as well as their parent compounds have also been tested by means of this fluorimetric assay. The proper fitting of the kinetic measurements is described by the parameters reported in Table 1.
:1 to 1:10. When the aggregation process was monitored by a turbidimetric assay (Fig. 4), TreRHQ and Tre(HQ)2 were able to significantly reduce the aggregation extent of an equimolar amount of Aβ1–42. At higher concentrantions of the tested compounds, all the Tre-HQ glycoconjugates were equally proficient in hampering the formation of both amorphous and fibrillar Aβ aggregates (Fig. 4). This ‘levelling effect’ in the turbidimetric assay is not observed when the formation amyloid-type fibrous aggregates was evaluated by means of the Thioflavin T (ThT) fluorimetric assay. ThT is an organic dye whose fluorescence intensity greatly enhances upon binding to amyloid fibrils both (Fig. S5†) in vitro and ex vivo. For this reason, the aggregation of Aβ1–42 in vitro has been monitored by measuring the fluorescence emission of ThT co-incubated with the amyloid peptide. TreHQ, TreRHQ and Tre(HQ)2, as well as their parent compounds have also been tested by means of this fluorimetric assay. The proper fitting of the kinetic measurements is described by the parameters reported in Table 1.
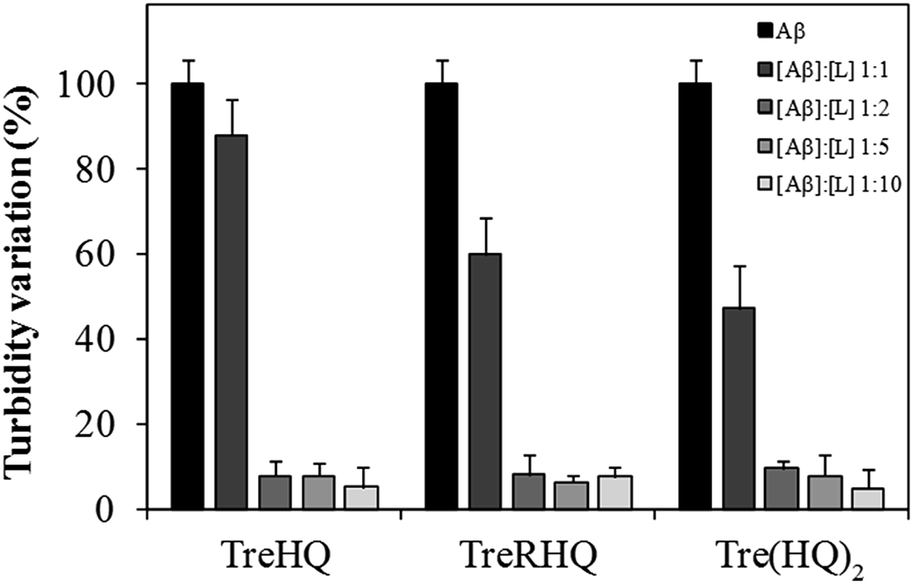 | ||
| Fig. 4 Turbidimetric measurements of Aβ1–42 alone (CTRL) or with each tested compound (TreHQ, TreRHQ and Tre(HQ)2. The peptide-to-ligand molar ratio ([Aβ]/[L]) ranged to 1:1 to 1:10. | ||
:1 to 1:10. The aggregation of Aβ alone is the control sample. All results are expressed as mean ± standard deviation (SD)
| Fmax − F0 (RFU) | K (h) | tlag (h) | Fmax − F0 (RFU) | K (h) | tlag (h) | |
|---|---|---|---|---|---|---|
| CTRL | ||||||
| 8.04 ± 0.06 | 1.84 ± 0.05 | 10.9 ± 0.3 | ||||
| [Aβ]/[L] | TreHQ | TreRHQ | ||||
| 1/1 | 6.0 ± 0.1 | 1.2 ± 0.1 | 12.2 ± 0.2 | 6.09 ± 0.08 | 1.79 ± 0.09 | 14.8 ± 0.3 |
| 1/2 | 5.3 ± 0.1 | 1.31 ± 0.06 | 11.9 ± 0.2 | 6.06 ± 0.04 | 2.55 ± 0.06 | 25.2 ± 0.2 |
| 1/5 | 5.0 ± 0.1 | 1.9 ± 0.1 | 18.6 ± 0.4 | 4.64 ± 0.05 | 1.46 ± 0.06 | 23.3 ± 0.2 |
| 1/10 | 4.9 ± 0.3 | 2.50 ± 0.07 | 20.0 ± 0.2 | 3.48 ± 0.04 | 2.88 ± 0.08 | 28.4 ± 0.3 |
| Tre(HQ)2 | (HQ)2 | |||||
| 1/1 | 7.7 ± 0.3 | 2.5 ± 0.5 | 11 ± 2 | 5.89 ± 0.04 | 2.24 ± 0.06 | 16.9 ± 0.4 |
| 1/2 | 5.1 ± 0.3 | 3.7 ± 0.5 | 12 ± 3 | 5.71 ± 0.06 | 2.18 ± 0.09 | 18.4 ± 0.1 |
| 1/5 | 3.6 ± 0.4 | 1.6 ± 0.9 | 14 ± 1 | 4.92 ± 0.08 | 2.12 ± 0.12 | 17.7 ± 0.4 |
| 1/10 | 1.9 ± 0.2 | 1.1 ± 0.5 | 16 ± 2 | 3.73 ± 0.07 | 1.60 ± 0.11 | 19.2 ± 0.2 |
| Tre | ||||||
| 1/1 | 7.0 ± 0.2 | 3.6 ± 0.3 | 14 ± 2 | |||
| 1/2 | 6.4 ± 0.1 | 3.6 ± 0.2 | 16.0 ± 0.8 | |||
| 1/5 | 5.1 ± 0.2 | 2.7 ± 0.5 | 16 ± 1 | |||
| 1/10 | 4.7 ± 0.3 | 2.8 ± 0.4 | 20.8 ± 0.9 | |||
The characteristic amyloid aggregation of Aβ is witnessed by the sigmoid trend of the ThT fluorescence response.31 A lag phase of 12 h is also a reliable value on the basis of the experimental conditions that mainly influence it, namely buffer composition, ion strength, protein concentration and temperature. The level of amyloid-type aggregation is proportional to the total fluorescence gain (Fmax − F0); for this reason, whenever a compound is tested, the greater the discrepancy of the Fmax − F0 value to that reported for Aβ alone (8.04), the higher the anti- or pro-aggregant activity of that compound.
An overall look at the kinetic parameters (Table 1) shows that the dose-dependent investigation of TreHQ, Tre(HQ)2, TreRHQ and (HQ)2 gives common outcomes: Fmax − F0 increases as the concentration of the tested compound increases. The lag phase is lengthened upon increasing the compound concentration as well. The shared behavior towards the amyloid aggregation is a further proof of the well-known antiaggregant propriety of 8-hydroxyquinoline moiety.6
As for TreHQ, the extent of the peptide aggregation is significantly reduced when the Aβ/compound ratio is even 1:1.
TreHQ also delays the fibril formation, being the lag phase (12.2 h) increased with respect to that of the control sample (10.9 h). Doubling the concentration of this compound, there is only a slight effect on the maximum fluorescence variation (Fmax − F0), whereas the lag phase values definitely increase when TreHQ concentration is five- or ten-fold to that of the amyloid peptide.
TreRHQ also causes dose-dependent changes on the kinetic profiles of the amyloid aggregation (Fig. 5).
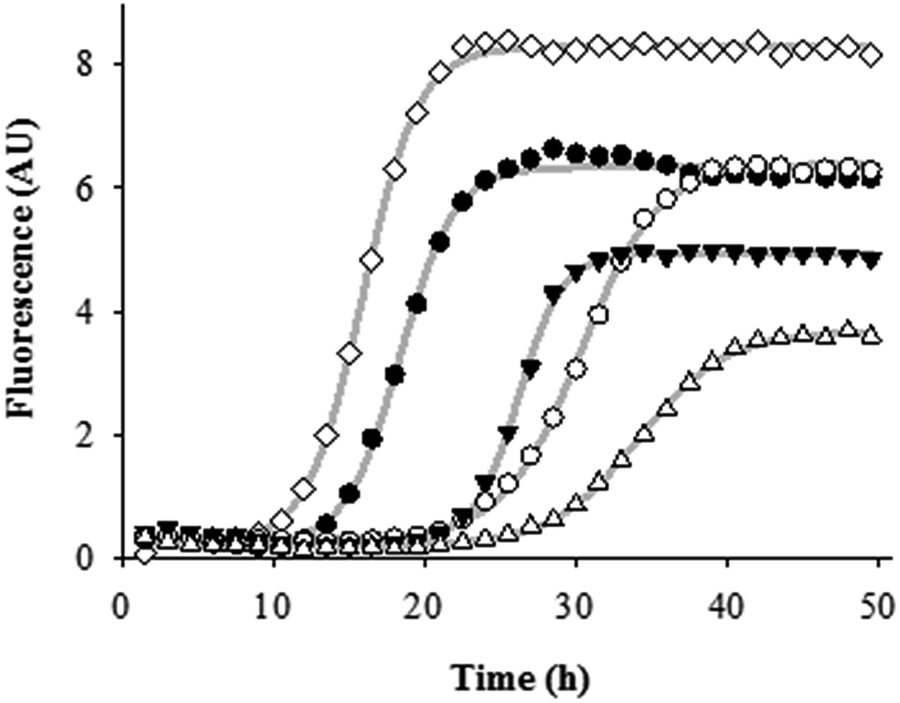 | ||
| Fig. 5 Sample kinetic profiles of Aβ1–42 aggregation (◊), in the presence of TreRHQ having different Aβ/compound ratios: 1:1 (●), 1:2 (○), 1:5 (▼), and 1:10 (△). Gray lines represent the fitted curves for each sample kinetic profile. | ||
Fmax − F0 is almost halved and the lag time is nearly doubled at the highest Aβ-to-compound ratio tested compared to fitted kinetic parameters of the Aβ aggregation alone. For these reasons, TreRHQ behaves better than TreHQ as antiaggregant agent, decreasing extent of amyloid aggregation and delaying formation of ThT-responsive aggregates.
Assaying Tre(HQ)2 on the aggregation of Aβ1–42 means testing the effect of two HQ moieties covalently linked to a Tre unit. This glycoconjugate undoubtedly reduces the amount of amyloid aggregates (Fmax − F0 decreases) in a direct proportion to the amount of Tre(HQ)2. Moreover, such a result is better than those produced by TreHQ and TreRHQ. However, differently by the mono-functionalized derivatives, Tre(HQ)2 had a poor effect on the lag phase of the amyloid aggregation process.
The quinoline derivatives HQ and (HQ)2, Tre and mixtures of Tre and HQ were also tested with the aim of getting information on the role that Tre unit or OHQ moiety could have on the antiaggregant property of the derivatives. The outcome (Tables 1 and S1†) reveals that both Tre and HQ have antiaggregant activity, although they are less effective than the derivatives in inhibiting Aβ aggregation. (HQ)2 also promotes the decrease of the Fmax − F0 value in a dose dependent manner, although the value of this parameter (3.73) is higher than that obtained in the presence of Tre(HQ)2 (1.9) at the highest peptide-to-compound ratio tested (1:10).
This clearly means that Tre has an active role on the antiaggregant activity of Tre(HQ)2 in keeping with the activity of Tre that progressively reduced the aggregation extent (Fmax − F0) with the increase of the concentration. As for the mixtures of Tre and HQ, the effect of Tre(HQ)2 is comparable to that for a combination of Tre and HQ in 1:2 molar ratio, whereas TreHQ has an activity higher than that of a Tre_HQ combination (1:1 molar ratio). Anyway, the main advantage of the covalent conjugation of HQ and Tre to form TreHQ and Tre(HQ)2 is the enhanced water solubility with respect to HQ. Moreover, the potential physiological fate the covalent derivatives should be reasonably different to that of the simple combination of the parent compounds.
Overall, mono- and bis-HQ derivatives of Tre display antiaggregant activity towards the formation of Aβ-containing fibrils at micromolar concentrations. Both the quinoline and the Tre units play an important role on this pathologically significant pathway of Aβ1–42 and two HQ units linked to Tre exert a better effect on the inhibition process than that showed by the monofunctionalized TreHQ. In line with this trend, the difunctionalization of cyclodextrin with HQ was also more effective strategy to suppress the amyloid aggregation than the mono-derivatization one (Fig. 6).22 The IC50 value of Tre(HQ)2 (38.9 μM) makes this compound a potential candidate for treating AD.
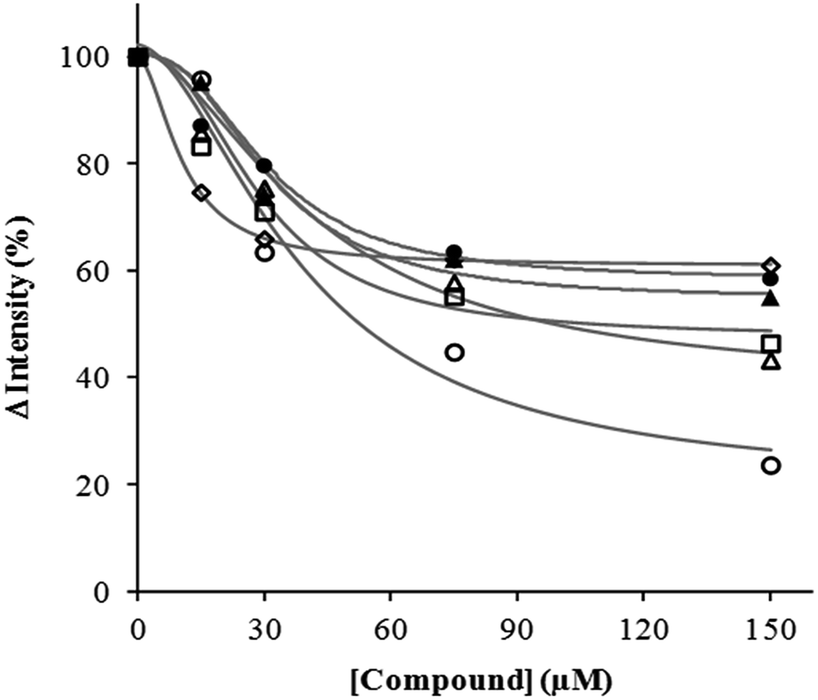 | ||
| Fig. 6 Dose-dependent variation of ThT-monitored Aβ1–42 aggregation in the presence of TreHQ (◊), TreRHQ (△), Tre(HQ)2 (○), (HQ)2 (□), Tre (●) and HQ (▲). Fitted curves related to any dose-dependent variation are also in the graph (gray lines). | ||
Molecular modeling
Molecular modeling was performed to visualize the interactions of Tre derivatives with Aβ1–42. Tre(HQ) and Tre(HQ)2 were the compounds of choice for this further study in order to compare two analogous systems that differ in the number of attached HQ moieties.The conformations of Aβ1–42 obtained from parallel tempering simulations highlight pliant domains, as also reported in previous contributions22,32,33 where short sections of alpha helix regions are stabilized along the backbone and all of them are connected through loop states.
Upon the binding of Tre(HQ) and Tre(HQ)2 to Aβ1–42, specific binding poses have been detected.
In particular, when Tre(HQ) is docked to Aβ1–42 three main binding poses with fairly close energy levels have been obtained. As similarly observed in the 8-hydroxyquinoline-appended cyclodextrin moiety docked to Aβ1–42,22 a first binding pose indicates a pi-stacking of the Y10 ring belonging to Aβ1–42 with the Tre(HQ) ligand, together with a ring–ring interactions between the α-glucose ring of the Tre and the aromatic ring of H14 (Fig. 7a), a non-covalent interaction observed in trehalose–histidine supramolecular frameworks.34 In the second binding pose the Tre(HQ) moiety lays among the intramolecular salt-bridges in the peptide scaffold of Aβ1–42 between the arginine group of R5 and the C-terminal carboxyl group of A42 (Fig. 7b). In the third binding poses, the Tre(HQ) ligand embraces the peptide scaffold of Aβ1–42 within the central loop region encompassing H14 and F20 residues (Fig. 7c).
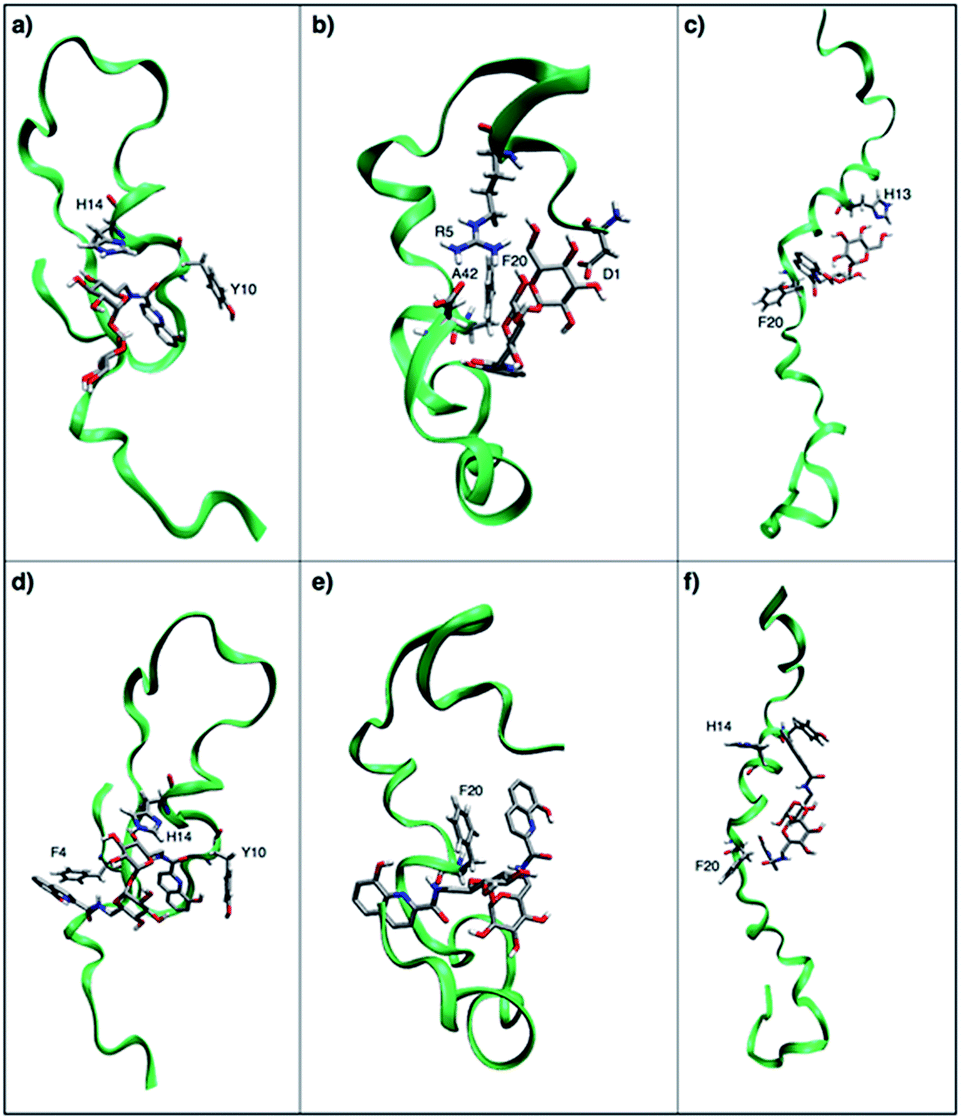 | ||
| Fig. 7 (a–c) The three lowest energy binding poses for the Aβ1–42/TreHQ complex. (d–f) The three lowest energy binding poses for the Aβ1–42/Tre(HQ)2 complex. Aβ1–42 sections are shown by green ribbons and the residues of Aβ1–42 interacting with TreHQ and Tre(HQ)2 are shown by solid sticks. Carbons are shown in silver, nitrogens are shown in blue and oxygens in red. The residue of Aβ1–42 involved in the binding site are labeled. | ||
The docking of Aβ1–42 with Tre(HQ)2 induces slightly different binding poses with fairly close energy levels. In particular, in the first binding pose the residues F4, H14 and F20 enclose the Tre(HQ)2 moiety, inducing a quite compact ligand-peptide supramolecular complex (Fig. 7d). In the second binding pose, F20 is stacked with one OHQ appended to the Tre ring (Fig. 7e), whereas in the third binding pose similarly to what found upon docking Tre(HQ) with the elongated cluster of Aβ1–42. Tre(HQ)2 lays in the central peptide loop encompassing the residues from H14 to F20 (Fig. 7f). The docking simulations provide a snapshot of molecular interactions to find out whether there is a direct interaction between Aβ and the compounds at atomic level. The results indicate that trehalose derivatives can bind to Aβ, suggesting a possible mechanism of interfering with the amyloid aggregation.
Conclusions
A new difunctionalized trehalose derivative was synthesized and characterized. Compared to the abundance of trehalose analogs and mimetics which have been studied as potential fungicides and antibiotics, few 6,6′-difunctionalized Tre derivatives have been designed and evaluated. In particular, Tre(HQ)2 represents the first example of 6,6′-difunctionalized trehalose derivative studied as an antiaggregant agent for self-induced Aβ aggregation. However, all trehalose derivatives have demonstrated a significant in vitro antioxidant capacity and antiaggregant ability. In particular, Tre(HQ)2 presents the highest capacity to scavenge free radicals suggesting that it is a powerful antioxidant with activity comparable to that of several polyphenols. Moreover, trehalose derivatives are able to strongly inhibit the peptide aggregation and the formation of amyloid fibrils whereas trehalose, 8-hydroxyquinolinecarboxamide (HQ) and bis(8-hydroxyquinoline) (HQ)2 alone are not as effective as the derivatives. Therefore, the conjugation of trehalose with 8-hydroxyquinoline induces synergistic effects that lead to superior antiaggregant properties. In particular, the 6,6′-difunctionalized compound results more effective than the corresponding 6-monofunctionalized suggesting that grafting two 8-hydroxyquinoline moieties on the disaccharide scaffold produces a better-performing antiaggregant compound at micromolar concentrations.Computational studies indicate that the derivatives can interact with Aβ stabilizing the monomer and thus avoiding the aggregation pathway. These finding that Tre-HQ conjugates, especially the 6,6′-difunctionalized compound, can interact with Aβ interfering with its self-aggregation are encouraging and underscore the potential of trehalose derivatives as therapeutics for amyloid-related pathologies.
Experimental
Materials
Reagents were purchased from common commercial suppliers and used without further purification. Tre was obtained from Sigma-Aldrich. 6,6′-Diamino-6,6′-dideoxy-α,α′-trehalose was obtained as reported elsewhere.356-Deoxy-6-[[(8-hydroxyquinoline)-2-carboxyl]-amino]-α,α′-trehalose (TreHQ) and 6-deoxy-6-[[(8-hydroxyquinoline)-2-methyl-amino]-α,α′-trehalose were synthesized as reported elsewhere.26
Thin layer chromatography (TLC) was performed on silica gel plates (Merck 60-F254) detecting carbohydrate derivatives by UV and/or anisaldehyde test.
Synthesis of 6,6′-dideoxy-6,6′-di[(8-hydroxyquinoline)-2-carboxyl]amino]-α,α′-trehalose
1,3-Dicyclohexylcarbodiimide (DCC, 118 mg, 0.57 mmol) was added to a stirred solution of 1-hydroxybenzotriazole hydrate (HOBt, 77 mg, 0.57 mmol) and 8-Hydroxyquinoline-2-carboxylic acid (108 mg, 0.57 mmol) in DMF. After 1 h, 6,6′-diamino-6,6′-dideoxy-α,α′-trehalose (90 mg, 0.26 mmol) was added and the mixture stirred at room temperature for a further 24 h. The solvent was then removed in vacuo and the remaining residue was purified by flash chromatography using a reversed phase column (RP-18, linear gradient H2O → EtOH).Yield: 75%. TLC: Rf = 0.45 (PrOH/AcOEt/H2O/NH3 5:3:1:2). (ESI-MS,+): m/z = 683.1 [M + H]+, 705.2 [M + Na]+. UV-vis (MOPS, pH 7.4): λ nm (ε M−1 cm−1) 253 (40100), 307 (3073), 351 (1913).
1H NMR (500 MHz, CD3OD) δ (ppm): 8.38 (d, 2H, J4,3 = 8.6 Hz, H-4), 8.16 (d, 2H, J3,4 = 8.6 Hz, H-3), 7.51 (t, 2H, J = 7.9 Hz, H-6), 7.41, (d, 2H, J5,6 = 7.7 Hz, H-5), 7.14 (dd, 2H, J7,6 = 7.5 Hz, J7,5 = 0.7 Hz, H-7), 5.08 (d, 2H, J1,2 = 3.8 Hz, Hs-1 of Tre), 4.08 (dt, 2H, J5,4 = 9.8 Hz, J5,6 = 4.9 Hz, Hs-5 of Tre), 3.81 (t, 2H, J = 9.3 Hz, Hs-3 of Tre), 3.75 (d, 4H, J6,5 = 4.9 Hz, Hs-6 of Tre), 3.46 (dd, 2H, J2,3 = 9.7 Hz, J2,1 = 3.8 Hz, Hs-2 of Tre), 3.22 (t, 2H, J = 9.3 Hz, Hs-4 of Tre).
13C NMR (125 MHz, CD3OD) δ (ppm): 167.3 (carbonyl Cs), 154.9 (Cs-8), 148.5 (Cs-2), 138.8 (Cs-4), 138.6 (Cs-9), 131.4 (Cs-10), 130.5 (Cs-6), 120.0 (Cs-5), 118.9 (Cs-3), 112.8 (Cs-7), 95.7 (Cs-1 of Tre), 74.3–72.2.2 (Cs-2, Cs-3, Cs-4 and Cs-5 of Tre), 41.1 (Cs-6 of Tre).
NMR spectroscopy
1H and 13C NMR spectra were recorded at 25 °C with a Varian UNITY PLUS-500 spectrometer at 499.9 and 125.7 MHz, respectively. The NMR spectra were obtained by using standard pulse programs from the Varian library. The 2D experiments (COSY, TOCSY, gHSQCAD, gHMBC) were acquired using 1k data points, 256 increments, and a relaxation delay of 1.2 s. The spectra were referred to the solvent signal.UV-visible spectroscopy and ESI-MS spectrometry
UV spectra were recorded on an Agilent 8452 A diode array spectrophotometer whereas ESI-MS experiments were performed on a Finnigan LCQ DECA XP PLUS ion trap spectrometer operating in the positive ion mode and equipped with an orthogonal ESI source (Thermo Electron Corporation, USA).Trolox equivalent antioxidant capacity assay
In vitro antioxidant assay was performed by 2,2′-azinobis(3-ethylbenzothiazoline-6-sulfonic acid) diammonium salt (ABTS) radical cation decolorization assay using 6-hydroxy-2,5,7,8-tetramethylchroman-2-carboxylic acid (Trolox) as reported elsewhere.26Aβ preparation
Aβ1–42 (Bachem) was dissolved in trifluoroacetic acid (TFA, 1 mg L−1) and sonicated for 10 min. TFA was removed by gentle streaming of argon. The peptide was then dissolved in 1,1,1,3,3,3-hexa-fluoro-2-propanol (HFIP) and incubated at 37 °C for 1 h. Following argon streaming, Aβ1–42 was dissolved again in HFIP, lyophilized, and then suspended in anhydrous dimethyl sulfoxide (DMSO, 200 μM) before the final dilution in the appropriate buffer for the aggregation assays.Aβ1–42 aggregation assay
Solutions containing Aβ1–42 (15 μM), ThT (60 μM) in phosphate buffer 10 mM at pH 7.4 were supplemented with the compounds of interest. The compound/peptide ratio ranged from 0 to 10. The solutions were incubated in a black 96-well plate (Nalge-Nunc, Rochester, NY) at 37 °C for 50 h in the Varioskan plate reader (Thermo Scientific). The kinetics of amyloid aggregation were followed by measuring the ThT fluorescence emission at 480 nm using excitation at 450 nm. All the measurements were carried out in triplicate and the experimental data were fitted by using the following equation:in which F0 and Fmax represent the initial and final fluorescence emissions of amyloid aggregation process, respectively; 1/k is the elongation rate constant and t1/2 the time at which the amplitude of ThT emission is 50% of the F0 − Fmax value. The lag time (tlag) is defined as the intercept between the time axis and the tangent of the curve with slope k from the midpoint of the fitted sigmoidal curve; this important parameter, according to the above definition, was calculated from the fitted parameters by using the following equation:
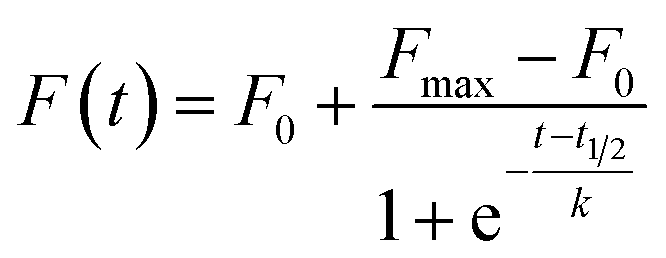
| tlag = t1/2 − 2k |
The kinetic parameters of any set of measurements were expressed as mean ± SD. In addition, point-based-ThT measurements were acquired to study the dose-dependent effect of the OHQ derivatives on the amyloid aggregation, as previously reported for similar compounds.22 Briefly, solutions containing Aβ1–42 (15 μm), ThT (45 μm) in phosphate buffer 10 mM at pH 7.4 were supplemented with the compounds of interest, being the compound/peptide ratio between 0 and 15. The solutions were incubated at 37 °C. After 50 h under shaking, ThT fluorescence intensity (450 nm excitation, 480 nm emission) was measured in triplicate. The percent variation between the fluorescence intensity of any sample and that of the control (ThT containing solution in the presence of Aβ1–42) was plotted as a function of the compound/peptide ratio and fitted by using the four parameter logistic nonlinear regression model:
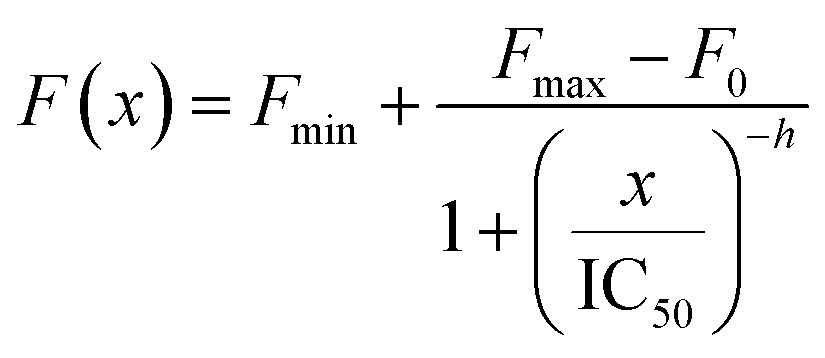
The kinetic measurements were also carried out in the absence of ThT. In this case, the amyloid aggregation was monitored by turbidimetric measurements. Positive and negative controls were used to ensure the validity of the results. Turbidity was calculated by the difference in absorbance at 405 nm between the sample and its matched control that did not contain Aβ.
Simulation details
The initial coordinates of Aβ1–42 were taken from the NMR coordinates reported elsewhere.36 (pdb code 1ZOQ). Those coordinates were considered in the zwitterion protonation states. The overall charge of the system was neutralized by adding three sodium ions. Parallel tempering (PT) simulations 15 ns long were run in explicit solvent with a total volume of 80 × 60 × 60 Å3, after an equilibration through 2 ns of MD in explicit solvent. We simulated 64 replicas distributed in the temperature range 300–500 K following a geometric progression. All replicas were simulated in NVT ensemble using a stochastic thermostat37 with a coupling time of 0.1 ps. A thermostat that yields the correct energy fluctuations of the canonical ensemble is crucial in parallel tempering simulations.38 Exchanges were attempted every 0.1 ps. The resulting average acceptance probability was 0.3 for all the replicas. The method of Daura and van Gunsteren39 was used in postprocessing phase to cluster the resulting trajectories, with a cutoff of 3 Å calculated on the backbone atoms as implemented in the clustering utility provided in the GROMACS package.40,41 From the former PT simulations we selected the main clusters of Aβ1–42 coordinates in order to run the docking simulations with the TreHQ and Tre(HQ)2 derivatives. Docking simulations have been performed through the DINC software.42Acknowledgements
The authors acknowledge support from the Consorzio Interuniversitario di Ricerca in Chimica dei metalli nei Sistemi Biologici (CIRCMSB), the Italian Ministero dell’Università e della Ricerca (FIRB RINAME).Notes and references
- F. M. LaFerla, K. N. Green and S. Oddo, Nat. Rev. Neurosci., 2007, 8, 499 CrossRef CAS PubMed.
- V. Chauhan and A. Chauhan, Pathophysiology, 2006, 13, 195 CrossRef CAS PubMed.
- C. Ballatore, V. M. Y. Lee and J. Q. Trojanowski, Nat. Rev. Neurosci., 2007, 8, 663 CrossRef CAS PubMed.
- R. León, A. G. Garcia and J. Marco-Contelles, Med. Res. Rev., 2013, 33, 139 CrossRef PubMed.
- A. A. Reinke and J. E. Gestwicki, Chem. Biol. Drug Des., 2007, 70, 206 CAS.
- H. LeVine, Q. Ding, J. A. Walker, R. S. Voss and C. E. Augelli-Szafran, Neurosci. Lett., 2009, 465, 99 CrossRef CAS PubMed.
- G. Vecchio and V. Oliveri, Chem.–Asian J., 2016 DOI:10.1002/asia.201600259.
- A. D. Elbein, Y. T. Pan, I. Pastuszak and D. Carroll, Glycobiology, 2003, 13, 17R CrossRef CAS PubMed.
- A. Arora, C. Ha and C. B. Park, FEBS Lett., 2004, 564, 121 CrossRef CAS PubMed.
- R. Liu, H. Barkhordarian, S. Emadi, C. B. Park and M. R. Sierks, Neurobiol. Dis., 2005, 20, 74 CrossRef CAS PubMed.
- J. Du, Y. Liang, F. Xu, B. Sun and Z. Wang, J. Pharm. Pharmacol., 2013, 65, 1753 CrossRef CAS PubMed.
- U. Krüger, Y. Wang, S. Kumar and E. M. Mandelkow, Neurobiol. Aging, 2012, 33, 2291 CrossRef PubMed.
- M. Tanaka, Y. Machida, S. Niu, T. Ikeda, N. R. Jana, H. Doi, M. Kurosawa, M. Nekooki and N. Nukina, Nat. Med., 2004, 10, 148 CrossRef CAS PubMed.
- S. Sarkar, S. Chigurupati, J. Raymick, D. Mann, J. F. Bowyer, T. Schmitt, R. D. Beger, J. P. Hanig, L. C. Schmued and M. G. Paule, Neurotoxicology, 2014, 44, 250 CrossRef CAS PubMed.
- D. Bini, A. Sgambato, L. Gabrielli, L. Russo and L. Cipolla, Biotechnology of Bioactive Compounds: Sources and Applications, 2015, p. 345 Search PubMed.
- J. Wang, B. Elchert, Y. Hui, J. Y. Takemoto, M. Bensaci, J. Wennergren, H. Chang, R. Rai and C. W. T. Chang, Bioorg. Med. Chem., 2004, 12, 6397 CrossRef CAS PubMed.
- Y. Igarashi, T. Mogi, S. Yanase, S. Miyanaga, T. Fujita, H. Sakurai and A. Ohsaki, J. Nat. Prod., 2009, 72, 980 CrossRef CAS PubMed.
- Y. L. Jiang, S. Miyanaga, X. Z. Han, L. Q. Tang, Y. Igarashi, I. Saiki and Z. P. Liu, J. Antibiot., 2013, 66, 531 CrossRef CAS PubMed.
- T. M. Ryan, B. R. Roberts, G. McColl, D. J. Hare, P. A. Doble, Q. X. Li, M. Lind, A. M. Roberts, H. D. T. Mertens, N. Kirb, C. L. L. Pham, M. G. Hinds, P. A. Adlard, K. J. Barnham, C. C. Curtain and C. L. Masters, J. Neurosci., 2015, 35, 2871 CrossRef CAS PubMed.
- A. I. Bush and R. E. Tanzi, Neurotherapeutics, 2008, 5, 421 CrossRef CAS PubMed.
- V. Oliveri, F. Attanasio, A. Puglisi, J. Spencer, C. Sgarlata and G. Vecchio, Chem.–Eur. J., 2014, 20, 8954 CrossRef CAS.
- V. Oliveri, F. Bellia, A. Pietropaolo and G. Vecchio, Chem.–Eur. J., 2015, 21, 14047 CrossRef CAS PubMed.
- V. Oliveri, M. Viale, C. Aiello and G. Vecchio, J. Inorg. Biochem., 2015, 142, 101 CrossRef CAS PubMed.
- C. Sgarlata, V. Oliveri and J. Spencer, Eur. J. Inorg. Chem., 2015, 5886 CrossRef CAS.
- V. Oliveri, F. Bellia and G. Vecchio, ChemPlusChem, 2015, 80, 762 CrossRef CAS.
- V. Oliveri, G. I. Grasso, F. Bellia, F. Attanasio, M. Viale and G. Vecchio, Inorg. Chem., 2015, 54, 2591 CrossRef CAS PubMed.
- V. B. Kenche, I. Zawisza, C. L. Masters, W. Bal, K. J. Barnham and S. C. Drew, Inorg. Chem., 2013, 52, 4303 CrossRef CAS PubMed.
- J. Bonda, X. Wang, G. Perry, A. Nunomura, M. Tabaton, X. Zhu and M. A. Smith, Neuropharmacology, 2010, 59, 290 CrossRef PubMed.
- A. Nunomura, R. J. Castellani, X. Zhu, P. I. Moreira, G. Perry and M. A. Smith, J. Neuropathol. Exp. Neurol., 2006, 65, 631 CrossRef CAS PubMed.
- A. Chan, J. Paskavitz, R. Remington, S. Rasmussen and T. B. Shea, Am. J. Alzheimer's Dis., 2008, 26, 571 CrossRef PubMed.
- G. I. Grasso, G. Arena, F. Bellia, E. Rizzarelli and G. Vecchio, J. Inorg. Biochem., 2014, 131, 56 CrossRef CAS PubMed.
- B. Teplow, N. D. Lazo, G. Bitan, S. Bernstein, T. Wyttenbach, M. T. Bowers, A. Baumketner, J. E. Shea, B. Urbanc, L. Cruz, J. Borreguero and H. E. Stanley, Acc. Chem. Res., 2006, 39, 635 CrossRef PubMed.
- A. Baumketner, S. L. Bernstein, T. Wyttenbach, G. Bitan, D. B. Teplow, M. T. Bowers and J. E. Shea, Protein Sci., 2006, 15, 420 CrossRef CAS PubMed.
- G. I. Grasso, G. Arena, F. Bellia, G. Maccarrone, M. Parrinello, A. Pietropaolo, G. Vecchio and E. Rizzarelli, Chem.–Eur. J., 2011, 17, 9448 CrossRef CAS PubMed.
- V. Cucinotta, A. Giuffrida, G. Grasso, G. Maccarrone, A. Mazzaglia, M. Messina and G. Vecchio, J. Sep. Sci., 2011, 34, 70 CrossRef CAS PubMed.
- S. Tomaselli, V. Esposito, P. Vangone, N. A. van Nuland, A. M. Bonvin, R. Guerrini, T. Tancredi, P. A. Temussi and D. Picone, ChemBioChem, 2006, 7, 257 CrossRef CAS PubMed.
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 014101 CrossRef PubMed.
- E. Rosta, N. V. Buchete and G. Hummer, J. Chem. Theory Comput., 2009, 5, 1393 CrossRef CAS PubMed.
- X. Daura, K. Gademann, B. Jaun, D. Seebach, W. F. van Gunsteren and A. E. Mark, Angew. Chem., Int. Ed., 1999, 38, 236 CrossRef CAS.
- B. Hess, C. Kutzner, D. van der Spoel and D. Lindahl, J. Chem. Theory Comput., 2008, 4, 435 CrossRef CAS PubMed.
- B. Hess, J. Chem. Theory Comput., 2008, 4, 116 CrossRef CAS PubMed.
- A. Dhanik, J. S. McMurray and L. E. Kavraki, BMC Struct. Biol., 2013, 13(suppl. 1), S11 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: ThT assay. See DOI: 10.1039/c6ra04204j |
| This journal is © The Royal Society of Chemistry 2016 |