
Graphene oxide with zinc partially substituted magnetite (GO–Fe1−xZnxOy) for the UV-assisted heterogeneous Fenton-like reaction†
Nor Aida Zubirab,
Julius Motuzasa,
Christelle Yacouac,
Xiwang Zhangd and
João C. Diniz da Costa*a
aFIM2Lab-Functional Interfacial Materials and Membranes Laboratory, School of Chemical Engineering, The University of Queensland, Brisbane, Qld 4072, Australia. E-mail: j.dacosta@uq.edu.au; Fax: +61 7 3365 4199; Tel: +61 7 3365 6960
bFaculty of Chemical Engineering, Universiti Teknologi MARA (UiTM), 13500 Pulau Pinang, Malaysia
cDepartment of Engineering, Université des Antilles, BP 250, 97157 Pointe à Pitre Cedex, Guadeloupe, France
dDepartment of Chemical Engineering, Monash University, Clayton Vic 3800, Australia
First published on 29th April 2016
Abstract
A series of graphene oxide (GO) and zinc partially substituted magnetite GO–Fe1−xZnxOy (0 ≤ x ≤ 0.285) catalysts were synthesised through a precipitation-oxidation method. The rate constants for the degradation of acid orange seven (AO7) proceeded at a significant faster rate under UV-irradiation (up to 670%) than the conventional heterogeneous Fenton-like reaction. The resultant catalysts were mesoporous, so there was no mass transfer limitation for AO7 to access active sites in the catalysts. Further, maximum increases of rate constant up to 220% occurred as the zinc molar concentration increased from x = 0 to x = 0.159. GO enhanced to incorporation of zinc into the combined metal oxide, whilst zinc limited crystal growth, thus forming smaller crystallite sizes. These features proved to be essential for the improved catalytic activity of the resultant catalysts. The optimised zinc molar value at x = 0.159 delivered the best catalytic activity.
1. Introduction
The heterogeneous Fenton-like reaction is of great interest for the degradation of persistent organic compounds using GO supported iron oxide nanocomposites, mainly due to their high surface area and decent catalytic properties.1–5 Compared to other types of iron oxide, magnetite (Fe3O4) offers higher catalytic activity with several interesting structural features.6,7 First, the presence of Fe2+ occupying on the octahedral site of Fe3O4 plays a key role as electron donor to initiate decomposition of hydrogen peroxide (H2O2) into hydroxyl radicals (HO˙) following the Haber–Weiss mechanism.7,8 Second, magnetite accommodates both Fe2+ and Fe3+ on the octahedral sites, allowing the Fe species to be reversibly oxidised and reduced while keeping the structure unchanged. Third, the Fe cations in the Fe3O4 structure can be substituted with different types of transition metal cations which can significantly affect the microstructure, physicochemical properties and catalytic activity of the resulting materials.9–11Moreover, recent studies have revealed that the isomorphic substitution of Fe3O4 with Co,12,13 Mn,9,14 Ti,15,16 Cr,17 and V15,18 have significantly enhanced its catalytic activity in various reaction. The exception is the substitution of Fe3O4 with Ni,19 which led to a decrease in catalytic activity as Ni2+ were mainly substituted with Fe2+ within the structure of Fe3O4. These variations were greatly dependent on the type of dopants, their concentration and occupancy of substitution sites which stimulates an effective generation of HO˙ radicals during catalysis. Nevertheless, the role played by GO in the UV-assisted Fenton-like reaction, when coupled with the heterogeneous Fenton-like catalyst (e.g. magnetite) and the photoluminescence catalyst (zinc oxide), is unknown. Therefore, this work investigates the influence of GO in the coupling with Fe3O4 and zinc oxide for the oxidative degradation of persistence organic compounds which remains unaddressed in the open literature.
This work shows for the first time the investigation of a series of zinc partially substituted magnetite with different concentrations being immobilised onto graphene oxide sheets as GO–Fe1−xZnxOy. Of particular interest, the effect of zinc on the physicochemical and catalytic properties of GO–Fe1−xZnxOy was systematically investigated by varying the zinc molar value 0 ≤ x ≤ 0.285. The catalytic performance of GO–Fe1−xZnxOy was evaluated using a model reaction of AO7 oxidative degradation in the UV-assisted and conventional heterogeneous Fenton-like reactions.
2. Experimental
FeCl3·6H2O (97%), FeCl2·4H2O (99%), Zn(NO3)2·6H2O (98%), NH4OH (30 wt% NH3), H2O2 (30%, w/w) and AO7 (orange II; 85%) were purchased from Sigma-Aldrich. All chemicals were of analytical grade and used as received except for graphite oxide which was prepared by modified Hummers's method.20,21 A series of composite GO/iron/zinc oxide catalysts with a zinc molar ratio (z) ranging from 0 to 0.4 were synthesised through a precipitation-oxidation method in the presence of graphene oxide (GO). Briefly, 62 mL of graphite oxide suspension (0.85 mg mL−1) was exfoliated under ultrasonication for 1 h to obtain an aqueous dispersion of GO. Typically for a sample z = 0.2, 8.64 mmol of FeCl3·6H2O, 4.32 mmol of FeCl2·4H2O and 0.86 mmol of Zn(NO3)2·6H2O were dissolved in 200 mL deionised water. After stirring for 30 min, this solution was heated to 90 °C and NH4OH solution was added dropwise into the heated solution until pH 4 was reached under constant stirring. This was followed by the gradual addition of GO suspension into the heated mixture and continuously stirred for another 30 min until homogeneously mixed. The iron and zinc precursor molar fractions were slightly changed for the preparation of samples with other z values.Subsequently, an appropriate amount of NH4OH solution was continuously added dropwise into the mixture until the pH reached a value of 11. The mixture was cooled to room temperature after being aged for 1 h at 90 °C under constant stirring. The resulting black precipitate was magnetically separated and washed three times with deionised water and ethanol and then dried at 60 °C for 48 h. For comparison, the same procedure was employed to synthesise zinc partially substituted magnetite with different zinc molar ratio (z = 0, 0.1, 0.2, 0.4) in the absence of GO solution.
The resultant materials were characterised by nitrogen sorption using a Tristar II 3020 (Micromeritics). The specific surface area and pore volume were determined using Brunauer–Emmett–Teller (BET) equation. The pore size distribution curves were calculated using non-local density functional theory (NLDFT), from the desorption branch of the isotherms. The XRD patterns were obtained using a Rigaku Smartlab X-ray diffractometer at 45 kV, 200 mA with a step size of 0.02° and speed of 4° min−1 using a filtered Cu Kα radiation (λ = 1.5418 Å). Surface analysis was performed on a Kratos Axis ULTRA X-ray photoelectron spectrometer (XPS) equipped with monochromatic Al Kα (hν = 1486.6 eV) radiation. The curve fitting was carried out using a Gaussian–Lorentz peak shape and Shirley background function. The binding energy was calibrated versus the carbon signal at 284.6 eV. The high resolution transmission electron microscopy (HRTEM) and scanning transmission electron microscope-energy dispersive X-ray spectrometer (STEM-EDS) elemental mappings were performed on JOEL 2010 operating at 200 kV, equipped with an energy dispersive X-ray (EDS) detector. Samples were prepared by placing a drop of diluted sample dispersion in ethanol onto a carbon-coated copper grid and air-dried prior to examination.
The catalytic performance of the catalysts was tested in the UV assisted heterogeneous Fenton-like reaction for the oxidative degradation of AO7. The experiments were carried out in a custom-made photo reactor equipped with 8 W UV-A lamps (Sylvania Blacklite F8 W/BL350, 330< λ < 370 nm) as described elsewhere.22 A 200 mL quartz tube was placed in the centre of the reactor, which was surrounded by the 4 UV-A lamps fitted to the wall of a cylindrical lead-line chamber in concentric arrangement. The distance of quartz tube was fixed at 10 cm from the UV lamps. In a typical experiment, 20 mg of the catalysts were added into the quartz tube consisting of 100 mL of 0.1 mM AO7 at initial pH solution of 3. The mixed suspension was kept under constant air bubbling for 30 min of dark adsorption. The reaction was initiated by turning on the UV lamps after the addition of H2O2 (22 mM) into the suspension. Sampling was carried out periodically at selected time interval. The collected suspension was then filtered through 0.2 μm Milipore syringe filters and immediately analysed. The concentration of AO7 was analysed by an Evolution 220 UV-Vis spectrophotometer (Thermo Fisher Sci.) at 484 nm. In order to compare the performance of the catalysts in the UV-assisted reactor, the control reaction was carried out in a conventional heterogenous Fenton-like reaction under same reaction conditions except of UV exposure.
3. Results and discussion
The synthesis of the starting catalysts was based on the variation of the zinc molar ratio (z). Table 1 displays the calculated values from XPS spectra to demonstrate the zinc molar value (x) actually incorporated into the mixed oxide. In this work, the general formula Fe1−xZnxOy for mixing oxides is used. In the case of oxygen, the notation (y) is used as the oxygen molar value is generally below 4 by mixing iron and zinc oxides. Table S1 (ESI†) shows that the Zn/Fe fractions for the GO–Fe1−xZnxOy samples are very close to the starting materials synthesis z values based on XPS results. Contrary to this, the Fe1−xZnxOy samples Zn/Fe fractions were much lower under the synthesis conditions used in this work. These results clearly indicate that GO played an important role to enhance Zn incorporation into the mixed oxides.| (z) starting Zn2+ | GO-Fe1−xZnxOy | Fe1−xZnxOy | ||
|---|---|---|---|---|
| (x) XPS for Zn2+ | (1 − x) XPS for Fe(2+,3+) | (x) XPS for Zn2+ | (1 − x) XPS for Fe(2+,3+) | |
| 0 | 0 | 1 | 0 | 1 |
| 0.1 | 0.090 | 0.910 | 0.001 | 0.009 |
| 0.2 | 0.159 | 0.841 | 0.020 | 0.980 |
| 0.4 | 0.285 | 0.715 | 0.167 | 0.833 |
Fig. 1a shows that the addition of GO was beneficial in terms of improving surface area, an important parameter in reaction engineering. For instance, GO–Fe1−xZnxOy surface areas (270–310 m2 g−1) were 68–130% larger than those of Fe1−xZnxOy (117–185 m2 g−1). The pore volumes for both samples (Fig. 1b) slightly decreased from 0.32 to 0.23 cm3 g−1 and 0.29 to 0.21 cm3 g−1 for Fe1−xZnxOy and GO–Fe1−xZnxOy as z was raised towards 0.4. As the actual amount of zinc x incorporated into the compounded increased concomitantly, there is a clear indication that increasing the amount of zinc led to denser structures. These results suggest the formation of different structures with and without GO. Fig. 1c and d show that both materials have pore size distribution (PSD) in the mesoporous region, though Fe1−xZnxOy has a much broader PSD aspect which is mainly associated with inter-particle space. In both cases, the PSD was narrowed as the zinc molar ration increased from 0 to 0.2.
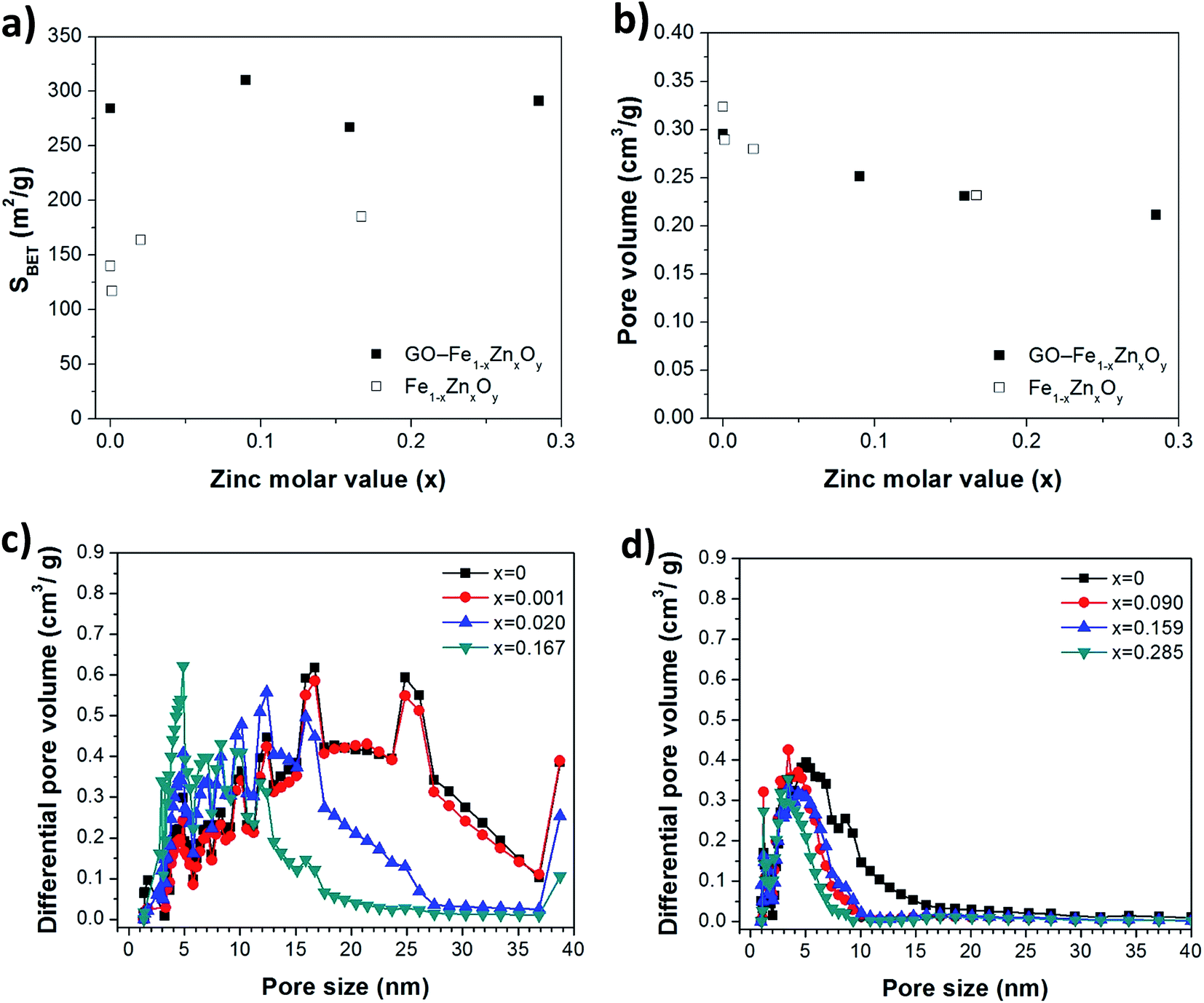 | ||
| Fig. 1 (a) BET surface area, (b) pore volume, and pore size distributions of (c) Fe1−xZnxOy and (d) GO–Fe1−xZnxOy. | ||
Further, GO–Fe1−xZnxOy consisted of more uniform and narrower PSD within the range of 1–10 nm (Fig. 1d). The addition of GO and in situ growth of Fe1−xZnxOy particles onto the GO sheets during the synthesis also induced the formation of micropores (<2 nm). This can be explained by the integration of a high aspect ratio two-dimensional (GO sheets) and zero-dimensional particles into a single material.2,23 By coupling the high surface areas and narrower PSD, the structural features of GO–Fe1−xZnxOy are therefore attributed to the intercalation of GO within the Fe1−xZnxOy particles.
The XRD patterns of GO–Fe1−xZnxOy are displayed in Fig. 2a. The diffraction peaks at the 2θ values of 30.2°, 35.6°, 43.3°, 53.6°, 57.4° and 63.1° were assigned to the (220), (311), (400), (422), (511) and (440) crystal planes of Fe3O4 with spinel structure (JCPDS no. 19-0629). A slight peak shift of (311) planes towards lower angle was observed with increasing the x values to 0.285. This suggests variations in the resultant crystal structure possibly due to the effect of zinc partial substitution into the Fe3O4 spinel structure. There was no visible secondary phase or impurity peaks, thus clearly confirming that zinc was isomorphically substituted into the Fe3O4 crystal structure, in good agreement with reports elsewhere.24,25
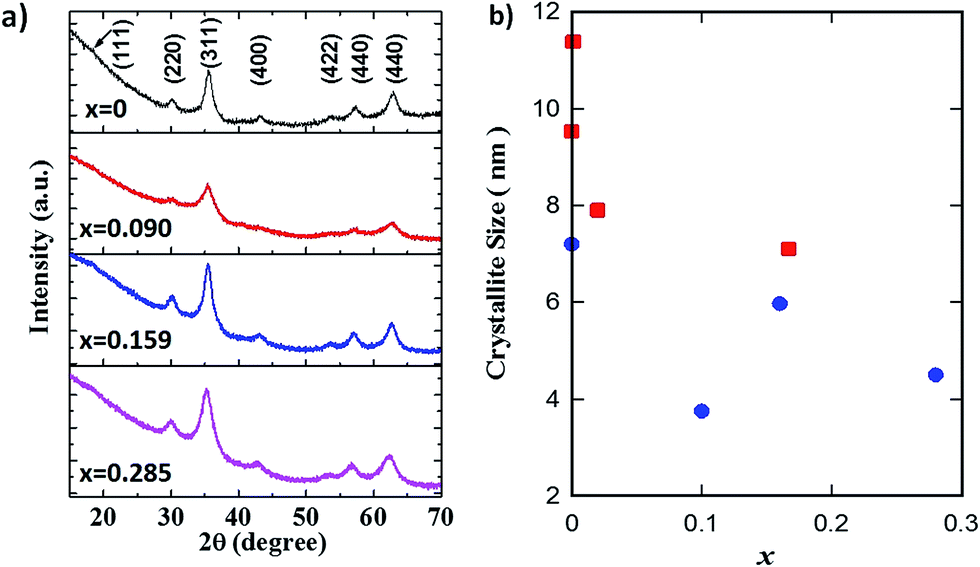 | ||
Fig. 2 (a) XRD patterns of GO–Fe3−xZnxOy, and (b) crystallite sizes of Fe1−xZnxOy ( ) NPs and GO–Fe1−xZnxOy ( ) NPs and GO–Fe1−xZnxOy ( ) samples at actual zinc molar values x. ) samples at actual zinc molar values x. | ||
Fig. 2b shows the crystallite sizes of the main peak at (311) planes of both samples calculated according to Scherrer's equation. Apart from a scatter for x = 0.1, the crystallite size of both samples reduced with increasing x. In principle, these results are indicating that zinc is limiting crystal growth, as the crystallite size reduced by 30% (10.08 to 6.99 nm) for Fe1−xZnxOy and by 40% (7.39 to 4.43 nm) for GO–Fe1−xZnxOy. In addition, the GO containing samples also resulted in smaller crystallite sizes ∼32% than non-GO samples, excluding the scatter variation of 56% for x = 0.1. These results therefore suggest that GO has further inhibited crystal growth. This could be attributed to the GO intercalation between particles, thus providing a hindrance effect of the particles to agglomerate and coalesce further.
The XPS analyses of GO–Fe1−xZnxOy in Fig. 3a and b confirm the spin–orbit doublet centred at 1021.7 and 1044.8 eV were corresponded to the Zn 2p3/2 and Zn 2p1/2 (except x = 0), respectively. The observed spin–orbit splitting between these two peaks was about 23 eV in line with those reported values of Zn2+ state.26–29 Fig. 3c displays the peaks centre at 711.1 and 724.6 eV in the high resolution Fe 2p scan which were assigned to Fe 2p3/2 and Fe 2p1/2 of Fe3O4.2,30–32 The peak of Fe 2p3/2 shift towards higher binding energy from 711.1 to 711.9 eV and became less intense as x increased. These differences can be explained by the changes in the electronic state of Fe and the Fe–O bond after zinc was partially substituted into Fe3O4.
 | ||
| Fig. 3 Wide scan XPS spectra of (a) high resolution spectra of (b) Zn 2p and (c) Fe 2p of GO–Fe1−xZnxOy at different zinc molar value x. | ||
Fig. 4 shows the HRTEM images of the GO–Fe1−xZnxOy. There were no significant differences in the morphology in both nanocomposites materials with x = 0 and x = 0.159. It was found that large amounts of particles were dispersed throughout the surface of GO sheets (Fig. 4a and c). From the magnified images in Fig. 4b and d, it was identified spherical particles with diameters of about 10–15 nm anchored on GO sheets. Furthermore, the inset HRTEM images revealed that the interplanar spacing of the lattice fringes of d = 0.25 nm, which corresponds well to the (311) planes of both Fe3O4 and Fe1−xZnxOy as measured at 2θ = 35.69° by XRD (Fig. 2). However, the latter slightly overlaps with the peak (101) at 2θ = 36.14° assigned to ZnO with interplanar spacing of d = 0.264 nm. The presence of zinc was examined by STEM-EDS elemental mapping. As displayed in Fig. 5a, b and d, e, the elements of Fe and O were detected and dispersed throughout the surface of both materials, while the zinc was only observed as white dots in Fig. 5f (GO–Fe1−xZnxOy, x = 0.159) but not in the sample without zinc as shown in Fig. 5c (GO–Fe1−xZnxOy, x = 0). Thus, these results further confirm the presence of zinc in GO–Fe1−xZnxOy.
 | ||
| Fig. 4 HRTEM images of GO–Fe1−xZnxOy at (a, b) x = 0 and (c, d) x = 0.159. | ||
 | ||
| Fig. 5 STEM images of GO–Fe1−xZnxOy with their corresponding EDS elemental mapping at x = 0 (a) Fe, (b) O and (c) Zn; and x = 0.159 (d) Fe, (e) O and (f) Zn. | ||
Oxidative degradation of AO7 in the presence of H2O2 was used as a model system to evaluate the catalytic activity of the synthesised samples. Initially, the samples were analysed for the conventional heterogeneous Fenton-like reaction in order to investigate whether the partial substitution of zinc was beneficial or detrimental towards the oxidative degradation of AO7 as displayed in Fig. 6. These results clearly indicate that altering the amount of zinc in the Fe1−xZnxOy did not affect their catalytic activity, which remained constant at ∼30% (±2%) as the x values varied from 0 to 0.167. However, a pronounced increase in the catalytic performance occurred when GO was incorporated as a catalyst support, forming GO–Fe1−xZnxOy. The removal efficiency of AO7 increased from 50% at x = 0 and peaked at 72% for x = 0.159, followed by a decline in removal efficiency to ∼52% for x = 0.285. The enhancement of the catalytic performance is clearly attributed to GO.
 | ||
| Fig. 6 The AO7 removal efficiency by (a) GO–Fe1−xZnxOy and (b) Fe1−xZnxOy at different zinc molar value (x) in the conventional heterogeneous Fenton-like. Experimental conditions: AO7 35 g L−1, H2O2 22 mM, catalyst 0.2 g L−1, T = 25 °C, pH 3 and 180 min of reaction. | ||
Further evaluation of the catalytic performance of the materials was carried out in the UV-assisted heterogeneous Fenton-like reaction. Fig. 7a displays representative UV-Vis spectra collected from an initial (0 min) to a final (120 min) testing condition. The disappearance of all peaks at 120 min of reaction strongly suggest the destruction of azo bond (–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–) in the chromophoric structure of AO7.33 It was found a relative good improvement from ∼30 to 52% of the catalytic activity for the Fe1−xZnxOy (x = 0.02) catalyst as the reaction was switched from the conventional to the UV-assisted. These results show that zinc has improved the UV-assisted catalytic activity of the compound without GO, though the AO7 degradation efficiency is relatively low. However, a complete new set of results was observed for GO–Fe1−xZnxOy. Fig. 7b shows a significant enhancement in efficiency within the first 60 min UV-irradiation as the AO7 degradation increased to 65% (x = 0) and peaked at 80% (x = 0.159). This clearly confirms the photoluminescence effect of zinc, leading to a faster catalytic activity for the partially substituted zinc catalysts. Further, the AO7 degradation went almost to completion at ∼98% at 120 min for all samples tested under UV-assisted conditions.
N–) in the chromophoric structure of AO7.33 It was found a relative good improvement from ∼30 to 52% of the catalytic activity for the Fe1−xZnxOy (x = 0.02) catalyst as the reaction was switched from the conventional to the UV-assisted. These results show that zinc has improved the UV-assisted catalytic activity of the compound without GO, though the AO7 degradation efficiency is relatively low. However, a complete new set of results was observed for GO–Fe1−xZnxOy. Fig. 7b shows a significant enhancement in efficiency within the first 60 min UV-irradiation as the AO7 degradation increased to 65% (x = 0) and peaked at 80% (x = 0.159). This clearly confirms the photoluminescence effect of zinc, leading to a faster catalytic activity for the partially substituted zinc catalysts. Further, the AO7 degradation went almost to completion at ∼98% at 120 min for all samples tested under UV-assisted conditions.
 | ||
| Fig. 7 The AO7 removal efficiency by GO–Fe1−xZnxOy in (a) UV-Vis spectra and (b) the UV-assisted heterogeneous Fenton-like reaction. | ||
Further analyses of results suggest that the oxidative degradation of AO7 using GO–Fe1−xZnxOy fitted well the pseudo-first-order reaction kinetics (R2 > 0.93). The rate constants in Fig. 8 clearly show that the AO7 degradation proceeded at a faster rate under UV-irradiation than the conventional heterogeneous Fenton-like reaction. The rate constants in Fig. 8 clearly show that the AO7 degradation proceeded at a faster rate under UV-irradiation than the conventional heterogeneous Fenton-like reaction, with significant increases in the order of 378 to 670%. Further maximum increases of rate constant 220% occurred as the zinc molar concentration increased from x = 0 to x = 0.159. These results are significant and clearly indicate that the photo-Fenton degradation of AO7 occurs at a much faster pace by combining zinc partially substituted magnetite with GO. Further, it is also noteworthy the improved photo-response of GO–Fe3O4 (GO–Fe1−xZnxOy, x = 0) under UV-irradiation, which is mainly attributed to GO.
 | ||
| Fig. 8 Rate constant (k) for AO7 removal efficiency by GO–Fe1−xZnxOy in the conventional and UV-assisted heterogeneous Fenton-like reaction. | ||
The resultant catalysts delivered major improvements under the UV-assisted over the conventional heterogeneous Fenton-like reactions as evidenced by the results in Fig. 7 and 8. In principle, this is attributed to the larger surface areas ∼270–310 m2 g−1 of GO–Fe1−xZnxOy as compared to 117–185 m2 g−1 for Fe1−xZnxOy. Further, the crystallite growth was limited mainly by GO and marginally by zinc, thus forming smaller crystallite sizes which proved to be essential for the improved catalytic activity of the resultant-catalyst. It is noteworthy that the highest catalytic activity was found to be at zinc molar value of x = 0.159 for both non-UV and UV assisted heterogeneous Fenton-like reaction (Fig. 8). There are no significant variations in surface areas (Fig. 1a) as a function of zinc molar value (x) whilst there is a small reduction in pore volume (Fig. 1b). However, the resultant samples are mesoporous (Fig. 1c and d) in nature, so there is no mass transfer limitation for AO7 to access active sites in the catalysts. Therefore, the catalytic activity peaking at x = 0.159 may be associated with the morphological change in the reduction of the crystallite size (Fig. 2b) coupled with the optimised zinc and magnetite molar ratio.
In order to explain the superior performance of the GO–Fe1−xZnxOy under the UV-assisted heterogeneous Fenton-like reaction, we propose a schematic of the reaction pathway as presented in Fig. 9. When the semiconductor catalyst is irradiated by UV light, a pair of photo-generated electrons holes are generated.34,35 In the presence of GO as the catalyst support, the holes are likely to be transferred from the valence band (VB) of the catalyst (Fe1−xZnxOy) to the highest occupied molecular orbital (HOMO) of graphene oxide attributed to the lower catalyst's VB position as compared to GO's HOMO.36 Meanwhile, the photo-generated electrons can only stay at the conduction band (CB) of the catalyst and participates in the surface catalysis to form radicals because of the CB position is also lower than the lowest unoccupied molecular orbital (LUMO) of GO.37 Owing to these reasons, GO played an important role in the UV-assisted Fenton-like reaction by inducing an effective charge separation during catalysis, supported by improved catalytic activity.
 | ||
| Fig. 9 Schematic of the UV-assisted heterogeneous Fenton-like reaction. | ||
4. Conclusions
A series of GO–Fe1−xZnxOy catalysts with varying zinc concentration were prepared through a precipitation-oxidation method in the presence of GO. The zinc partial substitution into the Fe3O4 was confirmed by the combined characterisation using XRD, XPS, and STEM-EDS analysis. There was no visible formation of secondary phase or impurity peaks, indicating the partial substitution of zinc into the Fe3O4 crystal structure with a good dispersion within the Fe3O4 matrix. The reaction kinetics peaked at zinc molar value (x) of 0.159 for GO–Fe1−xZnxOy towards the AO7's oxidative degradation in both conventional and UV-assisted heterogeneous and Fenton-like reactions. This improvement was associated with GO enhancing the incorporation of zinc into the mixed oxide matrix, coupled with the morphological features such as the reduction of the crystallite size coupled with the optimised zinc to magnetite molar ratio. Nevertheless, it was found that GO was a major player as it significantly increased the oxidative degradation of AO7 and also the reaction kinetics, thus conferring faster regeneration of the Fenton active species in producing more HO˙ radicals for the degradation of AO7 under both heterogeneous and UV-assisted Fenton-like reactions.Acknowledgements
The authors acknowledge the facilities and the scientific and technical assistance of the Australian Microscopy & Microanalysis Research Facility at the Centre for Microscopy and Microanalysis, The University of Queensland. N. A. Zubir gratefully acknowledges the generous financial support from Ministry of Higher Education Malaysia (MOHE) and Universiti Teknologi MARA (UiTM) for her study leave. J. C. Diniz da Costa acknowledges support via the Australian Research Council Future Fellowship Program (FT130100405).References
- S. Guo, G. Zhang, Y. Guo and J. C. Yu, Graphene oxide–Fe2O3 hybrid material as highly efficient heterogeneous catalyst for degradation of organic contaminants, Carbon, 2013, 60, 437–444 CrossRef CAS.
- N. A. Zubir, C. Yacou, J. Motuzas, X. Zhang and J. C. Diniz da Costa, Structural and functional investigation of graphene oxide-Fe3O4 nanocomposites for the heterogeneous Fenton-like reaction, Sci. Rep., 2014, 4, 4594, DOI:10.1038/srep04594.
- N. A. Zubir, C. Yacou, X. Zhang and J. C. Diniz da Costa, Optimisation of graphene oxide–iron oxide nanocomposite in heterogeneous Fenton-like oxidation of Acid Orange 7, J. Environ. Chem. Eng., 2014, 2, 1881–1888 CrossRef CAS.
- Y. Dong, J. Li, L. Shi, J. Xu, X. Wang, Z. Guo and W. Liu, Graphene oxide-iron complex: synthesis, characterization and visible-light-driven photocatalysis, J. Mater. Chem. A, 2013, 1, 644–650 CAS.
- Z. L. Hua, W. Q. Ma, X. Bai, R. R. Feng, L. Yu, X. Y. Zhang and Z. Y. Dai, Heterogeneous Fenton degradation of bisphenol A catalyzed by efficient adsorptive Fe3O4/GO nanocomposites, Environ. Sci. Pollut. Res., 2014, 21, 7737–7745 CrossRef CAS PubMed.
- R. Matta, K. Hanna and S. Chiron, Fenton-like oxidation of 2,4,6-trinitrotoluene using different iron minerals, Sci. Total Environ., 2007, 385, 242–251 CrossRef CAS PubMed.
- R. C. C. Costa, M. F. F. Lelis, L. C. A. Oliveira, J. D. Fabris, J. D. Ardisson, R. R. V. A. Rios, C. N. Silva and R. M. Lago, Novel active heterogeneous Fenton system based on Fe3−xMxO4 (Fe, Co, Mn, Ni): The role of M2+ species on the reactivity towards H2O2 reactions, J. Hazard. Mater., 2006, 129, 171–178 CrossRef CAS PubMed.
- X. Liang, Z. He, Y. Zhong, W. Tan, H. He, P. Yuan, J. Zhu and J. Zhang, The effect of transition metal substitution on the catalytic activity of magnetite in heterogeneous Fenton reaction: In interfacial view, Colloids Surf., A, 2013, 435, 28–35 CrossRef CAS.
- X. Liang, Z. He, G. Wei, P. Liu, Y. Zhong, W. Tan, P. Du, J. Zhu, H. He and J. Zhang, The distinct effects of Mn substitution on the reactivity of magnetite in heterogeneous Fenton reaction and Pb(II) adsorption, J. Colloid Interface Sci., 2014, 426, 181–189 CrossRef CAS PubMed.
- Y. Zhong, X. Liang, W. Tan, Y. Zhong, H. He, J. Zhu, P. Yuan and Z. Jiang, A comparative study about the effects of isomorphous substitution of transition metals (Ti, Cr, Mn, Co and Ni) on the UV/Fenton catalytic activity of magnetite, J. Mol. Catal. A: Chem., 2013, 372, 29–34 CrossRef CAS.
- L. Menini, M. C. Pereira, L. A. Parreira, J. D. Fabris and E. V. Gusevskaya, Cobalt- and manganese-substituted ferrites as efficient single-site heterogeneous catalysts for aerobic oxidation of monoterpenic alkenes under solvent-free conditions, J. Catal., 2008, 254, 355–364 CrossRef CAS.
- R. Amrousse, A. Tsutsumi, A. Bachar and D. Lahcene, N2O catalytic decomposition over nano-sized particles of Co-substituted Fe3O4 substrates, Appl. Catal., A, 2013, 450, 253–260 CrossRef CAS.
- R. C. C. Costa, M. De Fátima Fontes Lelis, L. C. A. Oliveira, J. D. Fabris, J. D. Ardisson, R. R. V. A. Rios, C. N. Silva and R. M. Lago, Remarkable effect of Co and Mn on the activity of Fe3−xMxO4 promoted oxidation of organic contaminants in aqueous medium with H2O2, Catal. Commun., 2003, 4, 525–529 CrossRef CAS.
- H. W. P. Carvalho, P. Hammer, S. H. Pulcinelli, C. V. Santilli and E. F. Molina, Improvement of the photocatalytic activity of magnetite by Mn-incorporation, J. Mater. Sci. Eng. B, 2014, 181, 64–69 CrossRef CAS.
- X. Liang, Y. Zhong, S. Zhu, L. Ma, P. Yuan, J. Zhu, H. He and Z. Jiang, The contribution of vanadium and titanium on improving methylene blue decolorization through heterogeneous UV-Fenton reaction catalyzed by their co-doped magnetite, J. Hazard. Mater., 2012, 199, 247–254 CrossRef PubMed.
- S. Yang, H. He, D. Wu, D. Chen, X. Liang, Z. Qin, M. Fan, J. Zhu and P. Yuan, Decolorization of methylene blue by heterogeneous Fenton reaction using Fe3−xTixO4 (0 ≤ x ≤ 0.78) at neutral pH values, Appl. Catal., B, 2009, 89, 527–535 CrossRef CAS.
- X. Liang, Y. Zhong, H. He, P. Yuan, J. Zhu, S. Zhu and Z. Jiang, The application of chromium substituted magnetite as heterogeneous Fenton catalyst for the degradation of aqueous cationic and anionic dyes, Chem. Eng. J., 2012, 191, 177–184 CrossRef CAS.
- X. Liang, S. Zhu, Y. Zhong, J. Zhu, P. Yuan, H. He and J. Zhang, The remarkable effect of vanadium doping on the adsorption and catalytic activity of magnetite in the decolorization of methylene blue, Appl. Catal., B, 2010, 97, 151–159 CrossRef CAS.
- Y. Zhong, X. Liang, Z. He, W. Tan, J. Zhu, P. Yuan, R. Zhu and H. He, The constraints of transition metal substitutions (Ti, Cr, Mn, Co and Ni) in magnetite on its catalytic activity in heterogeneous Fenton and UV/Fenton reaction: From the perspective of hydroxyl radical generation, Appl. Catal., B, 2014, 150, 612–618 CrossRef.
- W. S. Hummers and R. E. Offeman, Preparation of Graphitic Oxide, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- Z. Xiong, L. L. Zhang and X. S. Zhao, Visible-Light-Induced Dye Degradation over Copper-Modified Reduced Graphene Oxide, Chem.–Eur. J., 2011, 17, 2428–2434 CrossRef CAS PubMed.
- X. Zhang, D. K. Wang, D. R. Schmeda Lopez and J. C. Diniz da Costa, Fabrication of nanostructured TiO2 hollow fiber photocatalytic membrane and application for wastewater treatment, Chem. Eng. J., 2014, 236, 314–322 CrossRef CAS.
- J. Su, M. Cao, L. Ren and C. Hu, Fe3O4–Graphene Nanocomposites with Improved Lithium Storage and Magnetism Properties, J. Phys. Chem. C, 2011, 115, 14469–14477 CAS.
- S. S. Pati and J. Philip, A facile approach to enhance the high temperature stability of magnetite nanoparticles with improved magnetic property, J. Appl. Phys., 2013, 113, 044314, DOI:10.1063/1.4789610.
- J. M. Byrne, V. S. Coker, E. Cespedes, P. L. Wincott, D. J. Vaughan, R. A. D. Pattrick, G. Van Der Laan, E. Arenholz, F. Tuna, M. Bencsik, J. R. Lloyd and N. D. Telling, Biosynthesis of zinc substituted magnetite nanoparticles with enhanced magnetic properties, Adv. Funct. Mater., 2014, 24, 2518–2529 CrossRef CAS.
- A. Prakash, S. K. Misra and D. Bahadur, The role of reduced graphene oxide capping on defect induced ferromagnetism of ZnO nanorods, Nanotechnology, 2013, 24, 095705, DOI:10.1088/0957-4484/24/9/095705.
- R. R. Prabhakar, N. Mathews, K. B. Jinesh, K. R. G. Karthik, S. S. Pramana, B. Varghese, C. H. Sow and S. Mhaisalkar, Efficient multispectral photodetection using Mn doped ZnO nanowires, J. Mater. Chem., 2012, 22, 9678–9683 RSC.
- G. Lu, S. Qiu, J. Liu, X. Wang, C. He and Y.-J. Bai, Enhanced Electrochemical Performance of Zn-Doped Fe3O4 with Carbon Coating, Electrochim. Acta, 2014, 117, 230–238 CrossRef CAS.
- M. Ahmad, S. Yingying, H. Sun, W. Shen and J. Zhu, SnO2/ZnO composite structure for the lithium-ion battery electrode, J. Solid State Chem., 2012, 196, 326–331 CrossRef CAS.
- A. Prakash, S. Chandra and D. Bahadur, Structural, magnetic, and textural properties of iron oxide-reduced graphene oxide hybrids and their use for the electrochemical detection of chromium, Carbon, 2012, 50, 4209–4219 CrossRef CAS.
- H. He and C. Gao, Superparamagnetic, Conductive, and Processable Multifunctional Graphene Nanosheets Coated with High-Density Fe3O4 Nanoparticles, ACS Appl. Mater. Interfaces, 2010, 2, 3201–3210 CAS.
- N. A. Zubir, C. Yacou, J. Motuzas, X. Zhang, X. S. Zhao and J. C. Diniz da Costa, The sacrificial role of graphene oxide in stabilising Fenton-like catalyst GO–Fe3O4, Chem. Commun., 2015, 51, 9291–9293 RSC.
- X. Zhang, G. Li, Y. Wang and J. Qiu, Oxidative decomposition of azo dye C.I. Acid Orange 7 (AO7) under microwave electrodeless lamp irradiation in the presence of H2O2, J. Hazard. Mater., 2006, 134, 183–189 CrossRef CAS PubMed.
- H. Zhang, G. Chen and D. W. Bahnemann, Photoelectrocatalytic materials for environmental applications, J. Mater. Chem., 2009, 19, 5089–5121 RSC.
- S. K. Kansal, M. Singh and D. Sud, Studies on photodegradation of two commercial dyes in aqueous phase using different photocatalysts, J. Hazard. Mater., 2007, 141, 581–590 CrossRef CAS PubMed.
- X. Bai, L. Wang and Y. Zhu, Visible photocatalytic activity enhancement of ZnWO4 by graphene hybridization, ACS Catal., 2012, 2, 2769–2778 CrossRef CAS.
- S. Q. Liu, B. Xiao, L. R. Feng, S. S. Zhou, Z. G. Chen, C. B. Liu, F. Chen, Z. Y. Wu, N. Xu, W. C. Oh and Z. D. Meng, Graphene oxide enhances the Fenton-like photocatalytic activity of nickel ferrite for degradation of dyes under visible light irradiation, Carbon, 2013, 64, 197–206 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra04068c |
| This journal is © The Royal Society of Chemistry 2016 |