
Fabrication and characterization of ferritin–chitosan–lutein shell–core nanocomposites and lutein stability and release evaluation in vitro†
Abstract
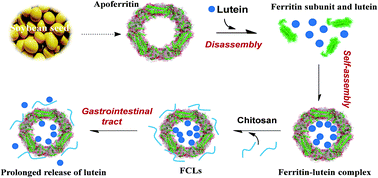
The application of bioactive lutein in the food industry is limited because of its poor water-solubility, instability, and low bioavailability. Nano-sized ferritin and chitosan provide a platform for fabricating shell–core system to encapsulate lutein. Herein, soybean seed ferritin (Glycine max) and chitosan stabilized lutein composites (FCLs) were fabricated by a unique reversible disassembly-reassembly character of apoferritin and ferritin–chitosan interaction. Results showed that lutein molecules were successfully encapsulated within the apoferritin with a molar ratio of 25.2 to 1 (lutein/ferritin), and the encapsulation efficiency and loading capacity were 16.8% and 2.50% (w/w), respectively. It was calculated that approximately 10 chitosan molecules were bound to a ferritin with a binding constant of 6.3 × 105 M−1, suggesting electrostatic interaction played an important role in ferritin–chitosan interaction. Results also indicated as much as 74.1% (w/w) of lutein was retained in FCLs after storage at 20 °C for 7 days. In addition, the photo- and heat-stability of lutein in FCLs were greatly improved as compared to free lutein. Furthermore, FCLs exhibited prolonged release of lutein in simulated gastrointestinal tract (GI) digestion as a result of ferritin coating and chitosan binding. Interestingly, food components exerted different effects in lutein release, EGCG, grape seed proanthocyanidin, and milk could inhibit while pectin could facilitate the release of lutein from FCLs. This work demonstrates an innovative strategy to solubilize, stabilize and control release of functional food nutrients.
 Please wait while we load your content...
Please wait while we load your content...

