
One step synthesis of silane-capped copper clusters as a sensitive optical probe and efficient catalyst for reversible color switching†
Abstract
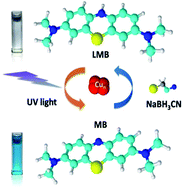
Discrete electron energy levels emerge in ultra small metal clusters due to a strong quantum confinement effect, resulting in some intriguing, unique and superior features such as photoluminescence and catalytic activity. In the present work, silane-capped copper clusters were synthesized by a facile one-pot synthetic protocol for the first time. Electrospray ionization mass spectrometry data shows that the metal core of the cluster is mainly composed of 4–5 copper atoms, and the results of X-ray photoelectron spectroscopy and Fourier-transform infrared spectroscopy indicate that the surface of the as-synthesized copper clusters is capped by a silane stabilizer. These few-atom copper clusters exhibit a dual-peak fluorescence feature, giving emission bands centered at 410 nm and 580 nm, respectively. In addition, the copper clusters also show excellent performance in chemo-sensing of hydrogen peroxide, linearly responding in a concentration range of 5–250 μM. More interestingly, a photo-reversible color switching system based on the redox reaction of methylene blue was built up by employing these Cu clusters as a catalyst. Reduction of methylene blue by Cu clusters' chemo-catalysis at ambient conditions and oxidation of leucomethylene blue by those clusters' photocatalysis under UV light irradiation lead to a recyclable colorless-blue switching effect within ∼3 minutes. The present work proves that the versatile silane-capped Cu clusters possess both molecular and semiconductor like properties, holding great promise in optical and catalytic application fields.
 Please wait while we load your content...
Please wait while we load your content...

