
FePt nanoparticles: a novel nanoprobe for enhanced HeLa cells sensitivity to chemoradiotherapy
Zhirong Bao†
a,
Mingyang He†b,
Hong Quan*a,
Dazhen Jianga,
Yanhong Zhengc,
Wenjing Qina,
Yunfeng Zhoud,
Feng Rena,
Mingxiong Guo*b and
Changzhong Jiang*a
aKey Laboratory of Artificial Micro- and Nano-Structures of the Ministry of Education and Center for Electronic Microscopy and Department of Physics, Wuhan University, Wuhan 430072, PR China. E-mail: 00007962@whu.edu.cn; czjiang@whu.edu.cn; Fax: +86 27 6875 3587; Tel: +86 27 6875 2567
bCollege of Life Sciences, Wuhan University, 430072 Wuhan, PR China. E-mail: guomx@whu.edu.cn
cOncology Department, Tongji Hospital, Tongji Medical College, Huazhong University of Science & Technology, PR China
dDepartment of Radiation and Medical Oncology, Zhongnan Hospital of Wuhan University, PR China
First published on 4th April 2016
Abstract
Chemoradiotherapy is a well-established treatment paradigm in oncology to consistently improve local tumor control compared to the sole administration of chemotherapy or radiotherapy. Despite its importance, few agents have been identified to further improve the therapeutic ratio and reduce the incidence of complications. Advances in nanomedicine have offered innovative strategies to improve chemoradiotherapy. In the present study, we evaluated the efficacy and safety of FePt nanoparticles (NPs) in chemoradiotherapy using HeLa cells and HEK293T cells. FexPt100−x NPs at different compositions (x = 26, 53 and 77) were synthesized and characterized by means of X-ray diffraction, transmission electron microscopy, and Fourier transform infrared spectroscopy. After ligand exchange, the cytotoxicity of FexPt100−x NPs was evaluated by MTT assay in HEK293T cells while the composition effects of FePt NPs on the cytotoxicity and the potential application of FePt NPs in combination with X-ray radiation at clinically relevant MV energies were investigated in HeLa cells in vitro. Besides, the cellular uptake of NPs was measured indirectly by atomic absorption spectroscopy and transmission electron microscopy. The results indicated that FexPt100−x NPs inhibited the growth of HeLa cells in a concentration- and composition-dependent manner after 24 h incubation, with low cytotoxicity to HEK293T cells at the given concentrations (0–20 μg mL−1). Furthermore, the combination of FePt NPs and radiotherapy resulted in a marked inhibition of HeLa cells, in contrast with that of the individual FePt NPs treated group or the radiation alone group. Moreover, Fe53Pt47 NPs, exhibiting significant cytotoxicity and enhanced radiosensitization effects on HeLa cells without damage to HEK293T cells, might theoretically satisfy the ultimate goal of personalized chemoradiotherapy. Our present work exhibited high therapeutic efficacy of FePt NPs in combination with radiotherapy without apparent cytotoxicity, suggesting the potential of FePt NPs as a promising nanoprobe in improving the outcome of tumor chemoradiotherapy.
Introduction
Cancer is one of the leading challenges in human healthcare today accounting for a high mortality rate worldwide.1,2 Although remarkable breakthroughs have been made in the development and application of highly efficient therapeutics to treat cancer, chemotherapy and radiotherapy as the two common approaches for clinic cancer treatment often fail owing to their own limitations. For example, the conventional anticancer drugs applied in chemotherapy generally lead to the insufficient tumor killing and severe side effects, due to the inevitable drug resistance, non-specific distribution and severe toxicity on normal cells.3–5 Complications (radiodermatitis, radiation pneumonia, etc.) induced by radiation inevitably arise when high prescription dose is essential to provide adequate dose deposition to tumors.6–8 Therefore, developing new and effective therapeutic means which may overcome the shortages of the single traditional therapeutic regime is extremely urgent and desirable. Driven by this need, chemoradiotherapy,9–12 the concurrent administration of chemotherapy and radiotherapy, has been developed to be a well-established treatment paradigm for treating cancers in oncology. Compared to either sequential treatment or sole implementation of chemotherapy or radiotherapy, chemoradiotherapy has been shown to combine the advantages of each therapy and consistently improve local tumor control and curative rates. Despite the dominant advantages in combination therapy, few agents have been identified to satisfy the need of chemoradiotherapy.With the developments of nanotechnology in biology and medicine over the past several decades, nanomaterials are anticipated to put forward effective strategies to achieve the aforementioned goals and improve chemoradiotherapy.13–15 Due to their unique surface chemistry and biocompatibility, nanoparticles (NPs) may preferentially accumulate in tumors by taking advantage of the enhanced permeability and retention (EPR) effect.16,17 This leads to a higher intratumoral drug concentration. Additionally, nanoparticles are unable to go through the tight junctions between endothelial cells on normal vascular linings, leading to a relatively low concentration in normal tissues surrounding the tumor. The NPs concentration differential between the tumor and normal tissue is an important prerequisite for a radiation sensitizer or anticancer drug because it should increase cellular radiation sensitivity or toxicity in tumors more than that of healthy tissue.14,18 Therefore, nanoparticles may be ideally suited for chemoradiotherapy to improve the therapeutic ratio and reduce the incidence of complications.
Recently, a variety of nanoparticles have been designed to improve chemoradiotherapy by serving for delivery of radiation sensitizing agents and/or chemotherapeutic drugs, such as the classical anticancer drugs,14,19–21 or nucleic acids.22–24 In addition to those nanoparticulate drug delivery systems, metal-based nanomaterials (e.g. Au, Ag)18,25–27 exist that can enhance the efficacy of radiotherapy because of their unique physical and chemical properties. Despite the efficacy in destroying cancer cells, these nanomaterials are by no means perfect due to their relatively larger size, complex preparation process and cumulative toxicity. Thus, the investigation to explore superior materials and methodology is still imperative. In the present study, we evaluate the possibility of FePt NPs to integrate chemotherapeutic with radiosensitization. FePt NPs have recently gained recognition as a superior material in cancer diagnosis and therapy due to the simple synthesis, excellent stability, biocompatibility and superparamagnetic property.28–30 Besides, the well-known anticancer drug based on the binding of platinum ions to DNA (e.g., cisplatin) encouraged us to evaluate the potential of FePt as an anticancer drug for chemotherapy.31,32 In addition to serving as chemotherapeutics, FePt NPs may also act as excellent radiosensitizers in cancer radiotherapy, based on the extremely high X-ray absorption of Pt.30 Therefore, FePt NPs could be ideal to combine the advantages of chemotherapy and radiotherapy, potentially improving the efficacy of tumor therapy and reducing the toxicity of nearby normal organs. Despite their high potential, there is currently no preclinical study evaluating the application of FePt NPs as a radiosensitizer and limited data overall exploring the use of this alloy nanoparticle for chemoradiotherapy. Furthermore, the inconsistencies in cytotoxicity may prevent their use as therapeutic agents in chemoradiotherapy.33
The aim of the present study was to evaluate the efficacy and safety of FePt NPs in chemoradiotherapy, and to assess some possible mechanisms of cytotoxicity and radiosensitizing effects of FePt NPs. To accomplish this goal, we designed and synthesized a series of FexPt100−x NPs (x = 26, 53 and 77), and then investigated the in vitro cytotoxicity in HEK293T and HeLa cells, intending to establish a systematic understanding about the composition effects of FePt NPs on the cytotoxicity. Moreover, viability assays were performed with to examine in vitro radiosensitization in HeLa cells while the effects of radiation on the cellular uptake of NPs were also evaluated via AAS. These findings may provide an important theoretical basis for the potential clinical application of FePt NPs as a novel nanoprobe for enhanced cancer cells sensitivity to chemoradiotherapy.
Experimental section
Materials
Chloroplatinic acid (H2PtCl6·6H2O, Reagent No. 1 Factory Of Shanghai Chemical Reagent Co., Ltd., Shanghai, China; analytical reagent), iron acetylacetonate (Fe(acac)3, Aladdin®, 98%), sodium borohydride (NaBH4, Sinopharm Chemical Reagent Co., Ltd, Shanghai, China, 96%) and cysteamine (Cys, Sigma, 95%) were used in this experiment. Other chemicals such as oleylamine, oleic acid, anhydrous ethanol, are all of analytical grade.Preparation of FePt NPs
In this paper we prepared three kinds of FePt nanoparticles with various alloying compositions by setting the molar ratios of Fe(acac)3 and H2PtCl6·6H2O as 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3, 1:1, 3:1 respectively. FePt NPs were synthesized using a chemical reduction method at a low temperature as previously described.34 Take the 1:1 as the example. Fe(acac)3 (0.386 mmol), oleyl amine (1.5 mL), oleic acid (1.5 mL) and anhydrous ethanol (100 mL) were mixed and stirred for 30 min. Then H2PtCl6·6H2O ethanol solution (19.3 mmol L−1, 0.386 mmol) was injected. After stirred for another half an hour, NaBH4 solution (200 mL, 65.7 mmol L−1) was gradually added and the reaction mixture was heated to 40 °C for 2 h before it was cooled down to room temperature. The product was precipitated by adding ethanol and separated by centrifugation. By repeating the dispersion–precipitation cycle, excess supernatant was discarded, and subsequently the product was dried and stored in vacuum conditions.
:3, 1:1, 3:1 respectively. FePt NPs were synthesized using a chemical reduction method at a low temperature as previously described.34 Take the 1:1 as the example. Fe(acac)3 (0.386 mmol), oleyl amine (1.5 mL), oleic acid (1.5 mL) and anhydrous ethanol (100 mL) were mixed and stirred for 30 min. Then H2PtCl6·6H2O ethanol solution (19.3 mmol L−1, 0.386 mmol) was injected. After stirred for another half an hour, NaBH4 solution (200 mL, 65.7 mmol L−1) was gradually added and the reaction mixture was heated to 40 °C for 2 h before it was cooled down to room temperature. The product was precipitated by adding ethanol and separated by centrifugation. By repeating the dispersion–precipitation cycle, excess supernatant was discarded, and subsequently the product was dried and stored in vacuum conditions.
Ligand exchange
The product obtained above (10 mg) and cysteamine (∼100 mg) were dissolved into ethanol in sequence. The mixture was sonicated for 6 h at 40 °C and then purified, dried, and stored as above mentioned for the synthesis of the FePt-Cys nanoparticles.Characterization techniques
The size and dispersity of the samples were observed on EM2010FEF-Ω transmission electron microscope (TEM, JEOL, Tokyo, Japan). Samples for TEM analysis were prepared by placing a drop of the sample suspension on a copper grid coated with carbon membrane. The structures of the samples were identified by XRD on a Bruker D8 Advance-X-ray Diffractometer (Bruker Corporation, Billerica, MA, USA) with Cu Kα radiation (λ = 1.5406 Å). The ligand exchange was verified with FTIR spectrophotometer (Thermo Nicolet5700) with a resolution of 4 cm−1. The ratio and concentrations of Fe and Pt were studied by atomic absorption spectroscopy (AAS) (PerkinElmer Analyst 800).Cell culture
Viability testing was conducted using different cell lines including HEK293T (human embryonic kidney cells) and HeLa (human cervical cancer cells) via MTT assays. Both cell lines were purchased from the type culture collection of the Chinese academy of sciences, Shanghai, China. Various cell lines were cultured in RPMI-1640 medium and Dulbecco modified Eagle's medium (DMEM) respectively, supplemented with 10% (v/v) fetal bovine serum (FBS, Gibco®, New York, USA), antibiotics penicillin (100 units per mL) and streptomycin (100 μg mL−1) (Beyotime Biotechnology, Shanghai, China) in a humidified atmosphere with 5% CO2 at 37 °C.Cytotoxicity assay of FexPt100−x NPs
The cytotoxicity of FexPt100−x NPs was evaluated by MTT assay using HEK293T cells. The medium containing FexPt100−x NPs was added in a dilution series, with final concentrations of 5, 10, 15 and 20 μg mL−1. The control well was a culture medium with no nanoparticles. After incubated with FexPt100−x NPs for 24 h and 72 h, 10 μL of MTT solution (5 mg mL−1 in PBS) was added to wells and cells further incubated at 37 °C for 4 h in a 5% CO2 humidified incubator. The medium was then carefully removed and the colored formazan were dissolved in 150 μL dimethylsulfoxide (DMSO). Finally, the absorbance of each well was quantified at 490 nm using a microplate reader (Thermo Fisher Scientific Inc., USA). The relative cell viability (%) related to control wells containing cell culture medium without nanoparticles was calculated by [A]test/[A]control × 100%.The inhibition of HeLa cells shared the procedure except the cell culture mentioned above and the incubation time. For further study, we adjusted the concentrations of different alloying compositions of FePt NPs to guarantee the same Fe concentrations to investigate the possible mechanism accounting for FePt NPs killing cells. Here, as a result, the corresponding Pt concentrations differed among the FexPt100−x NPs.
Inhibition effects of FexPt100−x NPs combined with radiotherapy on HeLa cells
According to the procedure above, HeLa cells were incubated with FexPt100−x NPs at low concentrations (less than10 μg mL−1) and then irradiated with 0, 2, 4, 6 and 8 Gray (Gy) respectively by the Varian Clinac IX Linear Accelerate (Zhongnan Hospital of Wuhan University, Wuhan, Hubei, China) with 6 MV photon beam. Incubated for 72 hours, the cell viability was measured by the MTT assay.Cellular uptake of FexPt100−x NPs with X-rays radiation
The cellular uptake of FexPt100−x NPs with X-rays radiation was quantified and performed in triplicate in the following manner. HeLa cells were cultured in a 6-well plate and incubated with 20 μg mL−1 FexPt100−x NPs and then irradiated with 0, 2, 4, 6 and 8 Gy X-rays respectively. After 24 h, the cells were washed three times with PBS, trypsinized, counted, and digested in hydrochloric acid (69%). Subsequently, the solution was sonicated and dispersed in deionized water. The intracellular Pt concentration was measured by AAS.Intracellular distribution of FexPt100−x NPs
Evidence of FexPt100−x NPs uptake was directly visualized by transmission electron microscopy (TEM). HeLa cells were treated with 20 μg mL−1 NPs for 24 hours. After the medium removed, cells were successively fixed with 2.5% glutaraldehyde and 1% osmic acid. Subsequently, a series of dehydration were performed and the cells were then embedded and sectioned by a diamond knife on a Leica ultramicrotome (Leica Microsystems, Wertzlar, Germany). TEM (H-7000FA, Hitachi, Japan, 100 kV) was used to observe the intracellular distribution of various nanoparticles.Results
Characterization of FePt NPs
Fe and Pt precursors typically play a crucial role in controlling the compositions of chemically synthesized nanoparticles.28 During the synthesis, the composition of the FexPt100−x nanoparticles was tuned by simply varying the stoichiometric ratio of the Fe and Pt precursors, where the value of x was found to vary from 26 to 77, as estimated from AAS analysis. Fig. 1A–C depicts the representative TEM micrographs of the FexPt100−x NPs along with the corresponding size distribution histograms, which were calculated from averaging the sizes of 100 nanoparticles. It could be seen that the average sizes of the corresponding constituent materials were all close to 3 nm with no apparent aggregation. Table 1 summarizes the size distribution and the corresponding composition of FexPt100−x NPs shown in Fig. 1A–C. The crystal structure of the nanoparticles characterized by XRD in Fig. 1D, showed that the four characteristic peaks are assigned to the (111), (200), (220), and (311) facet of the face-centered cubic (fcc) structure.35 During our synthesis, oleic acid (OA) and oleylamine (OAm) were chosen as the surfactants to stabilize the solution and prevent aggregation while the nanoparticles shielded with OA and OAm could hardly be dispersed in PBS buffer or deionized water and tend to precipitate within a few minutes. Thus, the nanoparticles were modified with cysteamine to improve the dispersion in PBS buffer. The existence of cysteamine capped on the surface of the FePt NPs was confirmed by FTIR spectra (Fig. 1E and F). In the spectrum of the bare FePt NPs, they all exhibited a broad band at around 3400 cm−1, corresponding to the N–H stretching of oleylamine. The peaks at 2920 cm−1 and 2850 cm−1 could be assigned to the symmetric and asymmetric stretching vibrations of –CH2 of OA/OAm while the peaks at 1560 cm−1 were responsible for the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching mode of oleic acid (Fig. 1E). After the cysteamine was conjugated onto the surface of FePt nanoparticles, the strong peaks around 3425 cm−1 and 1621 cm−1 could be attributed to the N–H stretching and bending vibration of cysteamine while the absorption at 3025 cm−1 was assigned to the C–H stretches (Fig. 1F). And it is worthy of noting that the S–H bond absorbed at 2540 cm−1 of cysteamine disappeared in the spectrum of FePt-Cys, implying the binding between mercapto groups and Pt atom due to the great affinity.36 These signatures indicate that cysteamine molecules were indeed anchored on the surface of the nanoparticles.
O stretching mode of oleic acid (Fig. 1E). After the cysteamine was conjugated onto the surface of FePt nanoparticles, the strong peaks around 3425 cm−1 and 1621 cm−1 could be attributed to the N–H stretching and bending vibration of cysteamine while the absorption at 3025 cm−1 was assigned to the C–H stretches (Fig. 1F). And it is worthy of noting that the S–H bond absorbed at 2540 cm−1 of cysteamine disappeared in the spectrum of FePt-Cys, implying the binding between mercapto groups and Pt atom due to the great affinity.36 These signatures indicate that cysteamine molecules were indeed anchored on the surface of the nanoparticles.
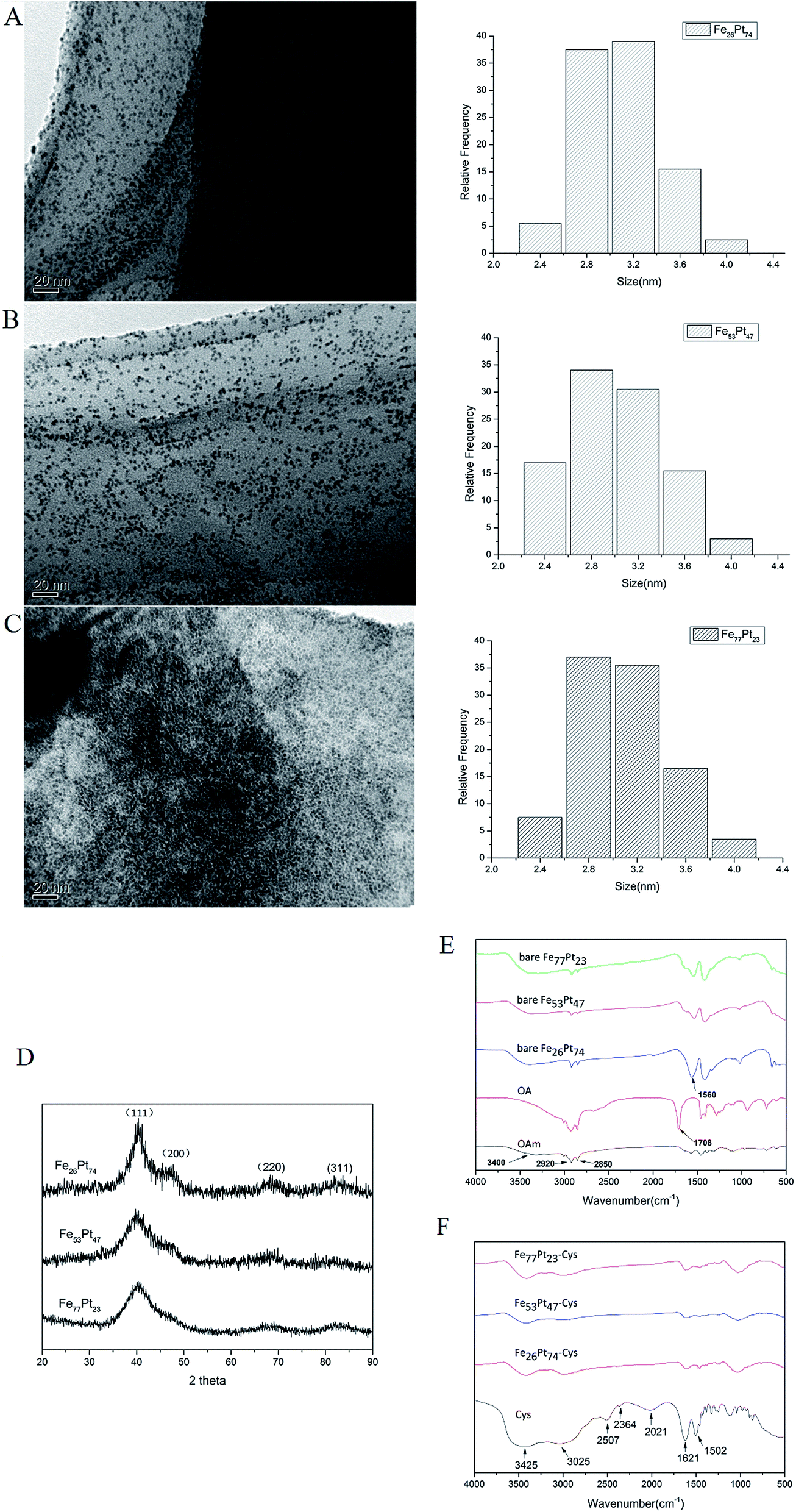 | ||
| Fig. 1 TEM, XRD characterization and the FTIR spectrum of FePt nanoparticles. The composition of the FexPt100−x nanoparticles was tuned by simply varying the stoichiometric ratio of the Fe and Pt precursors, where the value of x was found to be 26, 53 and 77, respectively, as calculated from AAS analysis. (A–C) TEM micrographs of the FexPt100−x nanoparticles along with the corresponding size distribution histograms. (A) Fe26Pt74, (B) Fe53Pt47, (C) Fe77Pt23 (D) XRD scans of FePt nanoparticles. (E) The FTIR spectrum of bare FePt nanoparticles, oleic acid (OA) and oleylamine (OAm). (F) The FTIR spectrum of cysteamine capped FePt nanoparticles and cysteamine. | ||
| Sample | Precursors and molar ratio | FexPt100−x | Size distribution (nm) | |
|---|---|---|---|---|
| 1 | Fe(acac)3/H2PtCl6·6H2O | 1:3 |
Fe26Pt74 | 3.08 ± 0.33 |
| 2 | 1:1 |
Fe53Pt47 | 3.01 ± 0.39 | |
| 3 | 3:1 |
Fe77Pt23 | 3.09 ± 0.36 | |
Cytotoxicity assay of FexPt100−x NPs
The in vitro cytotoxicity of water-solvable cysteamine capped FexPt100−x nanoparticles was evaluated in HEK293T cells by MTT assay. These FexPt100−x NPs were tested at concentration ranging from 0–20 μg mL−1 with incubation time of 24 h and 72 h (Fig. 2). The columns showed the mean survival fractions and the error bars represent the standard deviation. | ||
| Fig. 2 The in vitro cytotoxicity evaluated by MTT assay using HEK293T cells in the range of 0–20 μg mL−1 FexPt100−x NPs after different incubation time. (A) 24 h (B) 72 h. | ||
As shown in Fig. 2A, after 24 h of exposure, the MTT assay showed no significant cytotoxic response (the cell viability > 90%) detected at concentration below 20 μg mL−1 for the Fe53Pt47 and Fe77Pt23 nanoparticles. And even after prolonged incubation periods of up to 72 h, there is no detectable loss of cell viability (Fig. 2B), reflecting that FePt NPs have no significant cytotoxicity on HEK293T cells at concentrations of 20 μg mL−1 and below. In addition, Fe26Pt74 NPs displayed slightly stronger cytotoxic effects on HEK293T cells, with a ∼13% loss of cell viability at 20 μg mL−1 when incubated for 24 h.
In order to confirm the suppression of FePt NPs on tumor cells, we also detected the cell viability of HeLa cells. As shown in Fig. 3A, the explicit concentration-dependent effect was observed in FePt NPs, especially in the case of Fe53Pt47 NPs. By comparison, HeLa cells seemed to be more sensitive to the NPs than HEK293T cells. When HeLa cells were treated with Fe53Pt47 NPs, cell viability was dramatically decreased approximately from 93.1% to 71.2% with the concentration increased from 5 μg mL−1 to 20 μg mL−1. On the other hand, it seemed that various alloying compositions of FePt NPs showed different levels of toxic response at the same incubation intervals (Fig. 3A). Specifically, with the exposed dose to 20 μg mL−1, the cell viability in the case of Fe26Pt74, Fe53Pt47 and Fe77Pt23, was 53.2%, 71.2% and 84.4% separately. Obviously, Fe26Pt74 NPs displayed higher cytotoxic effects on HeLa cells than the other two samples. According to the viability testing, Fe77Pt23 NPs presented unconspicuous cytotoxicities over a dosage range of 0–20 μg mL−1.
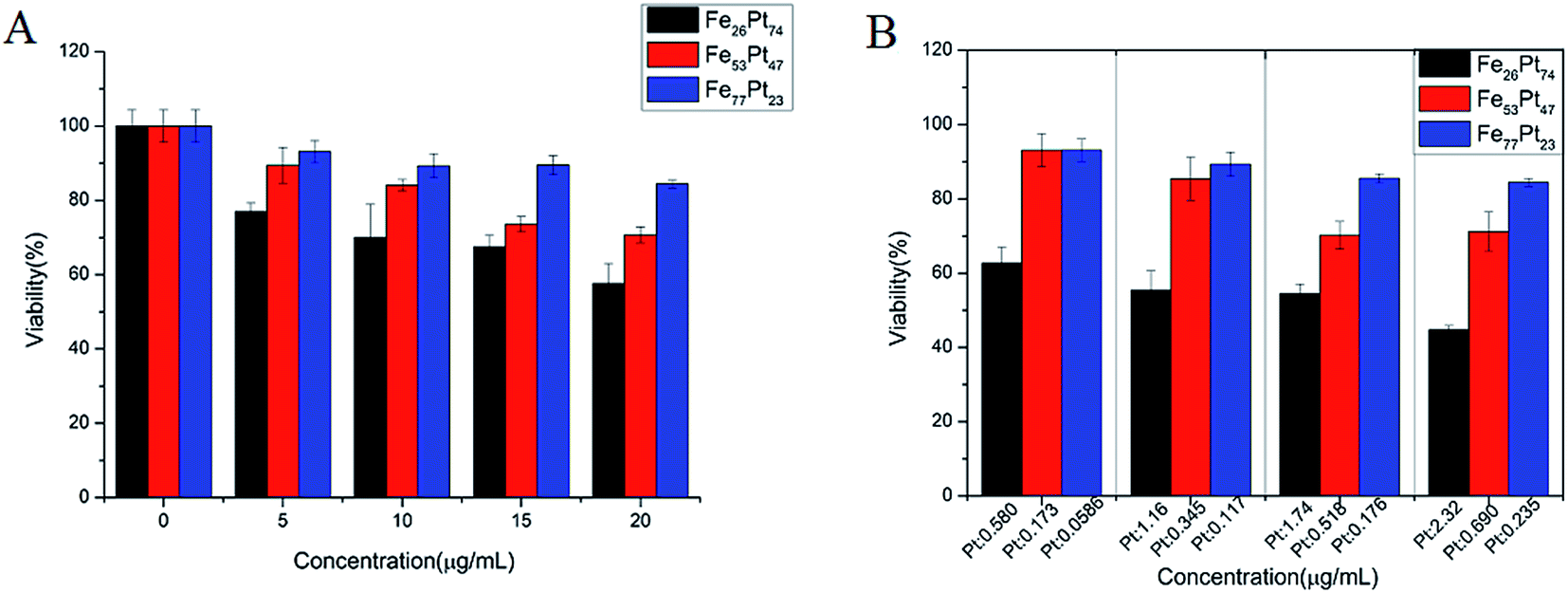 | ||
| Fig. 3 The in vitro cytotoxicity evaluated by MTT assay using HeLa cells after 24 h incubation with FexPt100−x NPs. (A) In vitro cytotoxicity of FexPt100−x NPs. (B) The contribution of Pt to the cytotoxicity. Herein, the concentrations of FexPt100−x NPs were adjusted to ensure the same Fe concentrations. Here, as a result, the corresponding Pt concentrations decreased among Fe26Pt74, Fe53Pt47 and Fe77Pt23 (B) were divided into four parts, with three columns in every part. (B) Pt concentrations decreased among Fe26Pt74, Fe53Pt47 and Fe77Pt23 in the three columns in every part. The data were normalized to controls (no particle exposure). Values were expressed as means ± standard deviation (SD). | ||
To further investigate the contribution of Fe or Pt to the cytotoxicity for future optimization of FePt NPs, we adjusted the concentrations of different alloying compositions of FePt NPs to guarantee the same Fe (or Pt) concentrations for MTT assay. Here, as a result, the corresponding Pt (or Fe) concentrations decreased (or increased) among Fe26Pt74, Fe53Pt47 and Fe77Pt23 at 37 °C for 24 h. As shown in Fig. 3B, FePt NPs incubated with cells for 24 h exhibited an obvious enhanced loss of cell viability with the increased Pt concentration. For instance, at the same Fe concentration of 0.228 μg mL−1, the cell viability presented as follows: 84.4% at 0.232 μg of Pt per mL of Fe77Pt23, 71.2% at 0.690 μg of Pt per mL of Fe53Pt47 and 44.7% at 2.32 μg of Pt per mL of Fe26Pt74. Nevertheless, with the increased Fe concentration, the cell viability showed irregular changes. Fe26Pt74 NPs still suffered greater loss of cell viability even at the relatively lower concentrations of Fe while Fe53Pt47 and Fe77Pt23 showed similar suppression effect. Hence, these results may reflect the fact that Pt presumably acts as a dominant role in the serious toxicity of FePt NPs, accounting for the stronger cytotoxicity of Pt rich nanoparticle.
Inhibition effects of FexPt100−x NPs combined with radiotherapy on HeLa cells
In order to assess the radiosensitization effects of FexPt100−x NPs, HeLa cells were cultured with or without FexPt100−x nanoparticles irradiated by serial doses of X-ray beams. As shown in Fig. 4A–D, without irradiation, the proliferation of HeLa cells was remarkably inhibited by FePt NPs while the cell viability was slightly reduced at higher concentrations, in the range of 1 to 10 μg mL−1. There was no significant difference in the toxicity of different formulations after 72 h incubation (Fig. 4A). On the other hand, without incubation with NPs, the percentages of viability showed a decrease at a higher radiation dose. When the NPs and X-ray beams were combined, results demonstrated that HeLa cells was significantly suppressed compared with that of radiation or NPs alone (Fig. 4B–D). For example, in terms of Fe53Pt47 NPs, the cell viability of the group decreased by ∼10% and ∼22% at the fairly low concentration 1 μg mL−1 combined with 2 Gy X-ray beams, compared with that of the group treated with 1 μg mL−1 NPs only or irradiated with 2 Gy alone, respectively (Fig. 4C). In addition, the NPs concentration and the radiation dose were both reduced in the NPs and radiation combination groups than that in the irradiation or NPs alone group. As displayed in Fig. 4C, the viability fractions of the group treated with 1 μg/mL Fe53Pt47 NPs and irradiated by 2 Gy X-ray beams showed no significant difference to that of the group only irradiated by 8 Gy X-ray beams or the groups treated with 10 μg mL−1 NPs only. Similar phenomenon could be observed in the other two kinds of nanoparticles. | ||
| Fig. 4 Inhibition of HeLa cells treated with FexPt100−x NPs combined with or without radiation. Incubation time was 72 h. (A) Without radiation (B–D) with various doses of X-rays radiation (0, 2, 4, 6 and 8 Gy). (B) Fe26Pt74 (C) Fe53Pt47 (D) Fe77Pt23. The data were normalized to controls (no particle exposure or radiation). Values were expressed as means ± standard deviation (SD). | ||
More interestingly, Fe53Pt47 NPs showed a slightly enhanced inhibition effect under radiation compared with the other two kinds of nanoparticles regarding any of the parameters studied. At concentration of 1 μg mL−1 with radiation dose of 4 Gy, for instance, the viability of HeLa cells incubated with Fe26Pt74, Fe53Pt47, Fe77Pt23 NPs was 54.9%, 47.0%, 59.5%, respectively (Fig. 4B–D).
Cellular uptake with X-rays radiation
For quantification of the influence of external beam radiation on the intracellular uptake of the particles, we seeded HeLa cells in 6 wells plates and incubated the cells with 20 μg mL−1 FexPt100−x NPs. After various doses of X-rays radiation (0, 2, 4, 6 and 8 Gy), the uptake amounts of nanoparticles was determined by AAS analysis after incubation for 24 h. The cellular Pt contents were plotted with the remaining of the measured data minus the background detection from the control (−5.6 ± 0.5 ng/1.0 × 105 cell).As seen from the intracellular uptake curves depicted in Fig. 5A, without radiation, the intracellular concentration of Pt were 28.6, 45.1, 34.7 ng/1.0 × 105 cell, after incubation with Fe26Pt74, Fe53Pt47 and Fe77Pt23 NPs respectively. When radiation was given following drug administration, the Pt contents, as expected, demonstrates apparent increase compared to the poor uptake without X-rays beams. These results support a dose-dependent mechanism for FexPt100−x uptake. Curiously, FexPt100−x NPs treatment resulted in a dramatically lower uptake when larger dose given to cells. Specifically, cells treated with Fe26Pt74 NPs show the unexpected sharp drop-off between 2 and 4 Gy. In contrast, the peak uptake of Pt in Fe53Pt47 NPs-treated cells was observed at dose of 6 Gy compared to the 4 Gy of Fe77Pt23 NPs treated cells.
 | ||
| Fig. 5 Cellular uptake and possible mechanism. (A) The influence of external beam radiation on the intracellular uptake of the particles. The cellular concentrations were plotted with the remaining of the measured data minus the background detection from the control (−5.6 ± 0.5 ng/1.0 × 105 cell). (B) TEM images of HeLa cells incubated with FexPt100−x NPs. (C) Illustration of a possible mechanism accounting for FePt NPs killing HeLa cells with or without radiation. After cellular uptake, FePt nanoparticles were oxidized to give Fe2+ and Pt2+ ions. The Pt2+ ions enter into the nucleus and lead to apoptosis of the HeLa cells. Under radiation, increased uptake of NPs tends to release more Fe2+ and Pt2+ ions and thus leads to enhanced cytotoxicity and radiosensitization effects. | ||
Intracellular distribution of FexPt100−x NPs
The intracellular distribution of cysteamine coated FePt NPs in HeLa cells is illustrated qualitatively via obtaining typical conventional TEM micrographs using a Phillips CM 100 TEM at 100 kV. As predicted from the cellular uptake detected quantitatively using AAS (Fig. 5A), a highly electron-dense material accumulation was observed in TEM images after 24 h exposure with the NPs at 20 μg mL−1 (Fig. 5B). Specifically, some nanoparticles were found near the cell membranes, large aggregates of nanoparticles were entrapped in the secondary lysosomal structures and the rest were distributed in the cytoplasm and nucleus regions, consistent with the previous studies.33,37–39 Fig. 5B showed that NPs attached to the cell membrane were undergoing the uptake process, which may imply the likely endocytotic pathway.Discussion
In the present work, we prepared FexPt100−x NPs with 3 nm in diameter and modified their surface with cysteamine to ensure the water solubility and further bioconjugation. A series of evaluations were performed to characterize the alloying compositions, structure, size distribution and surface chemistry of NPs. To assess the potential application of FexPt100−x NPs in chemoradiotherapy, MTT viability analysis was conducted on HeLa and HEK293T cells. The results indicated that NPs significantly suppressed the proliferation of HeLa cells (Fig. 3A), while exerting no significant cytotoxic effects on HEK293T cells (Fig. 2A) at the given concentration. Actually, such kind of cell-specific death response could be observed in other types of metal-based nanoparticles.40–42 For example, gold nanoparticles, even in the absence of any specific functionalization, could induce apoptosis in cancer cells while the tested normal cells remained unaffected.40 This may be contributed to the following reasons. (1) Different cell lines showed varying extent of nanoparticle uptake. Although nanoparticles cellular uptake and localization in cytoplasmic endosomes occurred in all cells, it has been demonstrated that nanoparticles show greater uptake in cancer cells than that in normal cells.43,44 This may be due to the unique pathophysiology of tumors, such as their enhanced permeability and the higher rate of uptake triggered by abnormally high metabolic rate. (2) The production of reactive oxygen species (ROS) caused by metal-based NPs may be considered as a mechanism for the specific killing of cancer cells.42,45,46 It is reported that cancer cells are characterized by higher levels of ROS and lower ROS survivability while normal cells have lower oxidative stress levels and are able to bear higher levels of ROS. As a result, once ROS levels are further elevated by metal-based NPs, the survivability of cancer cells is lower than that of normal cells and may be relatively susceptible to death. Although further studies are still required to explore the origin of such cell specific responses, what we would like to highlight is that there may be an interaction between FePt NPs and cellular and (or) subcellular receptors in certain cells. The understanding about the selective cancer-killing ability of nanoparticles may be an important step forward in nanomedicine.Furthermore, a reduction in viability of HeLa cells exposed to FePt NPs for 24 h was observed in concentration- and composition-dependent manners. Herein, Fe26Pt74 and Fe53Pt47 NPs exerted remarkable cytotoxic effects among the three samples while lower levels of cytotoxicity were observed for Fe77Pt23 NPs to HeLa cells at the given concentrations. Previously, Sun et al.47 suggested that FePt NPs did not display the composition-dependent relationship in cytotoxicity, presumably owing to the different synthesis method and surface coatings. As described above, Fe53Pt47 NPs may evolve to become the relatively safe and effective drugs for cancer treatment considering the observed potent cytotoxicity toward HeLa cells and the non-cytotoxic effects on HEK293T cells. Due to the concentration- and composition-dependent relationship in cytotoxicity, it required serious consideration to select nanoparticles with proper composition and concentration to achieve a maximum therapeutic effect in treating tumors. The optimal nanoparticles may intelligently suppress the proliferation of tumor cells without damaging the normal cells, and thus potentially improve the therapeutic efficacy in chemotherapy. To further investigate the contribution of Fe or Pt to the cytotoxicity for future engineering nanoparticles, we performed a series of control experiments to analyze the effects of Fe or Pt. As predicted from previous experiments,31,32 FePt NPs exhibited enhanced loss of cell viability with the increased Pt concentration (Fig. 3B), which presumably resulted from the mechanism that the release of Pt2+ ions increased for particles with high platinum content (e.g., Fe26Pt74). Similar to the well-known platinum-containing anticancer drugs (e.g., cisplatin, carboplatin, and oxaliplatin), Pt2+ ions could damage the DNA double-helix chains and eventually cause the apoptosis of tumor cells.31
To explore the potential applications of FePt NPs in chemoradiotherapy, HeLa cells were exposed to 6 MV photon beam with or without nanomedicine incubation. As shown in Fig. 4A–D, FexPt100−x NPs suppressed the proliferation of HeLa cells in a concentration-dependent manner in the NPs treated alone groups, while X-ray beams resulted in the inhibition of proliferation in a radiation dose-dependent manner in the irradiation alone groups. When the NPs and X-ray beams were combined, results demonstrated that the suppression ratios remarkably increased compared with that of radiation or NPs alone (Fig. 4B–D). More interestingly, low concentration of FePt NPs combined with low-dose X-ray beams exhibited efficient inhibition of HeLa cells growth in comparison with the results in the high-dose X-ray radiation or high-concentration of NPs alone group. In other words, both the NPs concentration and the radiation dose could be reduced when NPs and radiation were combined. Indeed, this factor could protect normal tissues from injury and increase the therapeutic efficacy of FePt NPs. Therefore, compared to the conventional treatments dependent only on therapeutic agents or X-ray beams for tumor inhibition, FePt NPs exhibited cytotoxicity and radiosensitization effects, which might lead to a new strategy to improve the therapeutic efficacy in chemoradiotherapy. Given the combined effect of NPs and radiation, one could employ proper concentration of nanoparticles to cooperate with different radiation treatment plans such as the conventional external radiotherapy treatment (conventional dose, 2 Gy/fraction) or hyperfractionated accelerated radiotherapy cases (a higher dose, e.g. 6 Gy/fraction).
Additionally, unlike the results of 24 h incubation, the cell viability of HeLa cells no longer demonstrated strong dependency on their compositions and concentration after 72 h incubation of NPs (Fig. 3A and 4A). This could be due to the drug resistance in residual cells.48–50 Generally, several factors exist that can lead to the drug resistance, including the poor absorption of administered drugs, increased drug excretion, increased repair of drug-induced damage, and decreased drug sensitivity due to genetic and epigenetic alterations. Hence, the residual cells grown in culture shown to possibly become resistant to cancer drugs, and thereby influence the results of MTT assays using FePt NPs for 72 h. Similar phenomenon could be commonly observed in other NPs systems.25,31 On the other hand, when combined with radiation, Fe53Pt47 NPs showed a slightly enhanced inhibition effect at same concentrations compared with the other two kinds of nanoparticles regarding any of the parameters studied (Fig. 4B–D). Taking the above results together, Fe53Pt47 NPs exhibiting significant cytotoxicity and enhanced radiosensitization effects on HeLa cells without damage to HEK293T cells, might theoretically satisfy the ultimate goal of personalized chemoradiotherapy.
Allowing for the importance of the uptake of NPs in nanotoxicology, AAS analysis was conducted to investigate the influence of external beam radiation on the intracellular uptake of the particles. As seen in Fig. 5A, after X-rays irradiation, the intracellular concentration of Pt content increased apparently compared to the group without irradiation. The enhanced cellular uptake of FePt NPs may be attributed to the specific response of the cell membrane to X-ray radiation. Due to the interaction between X-ray beam and biomolecules of the cell membrane, the permeability and integrity of cell membrane is destroyed.51,52 The irradiation-induced increases in cell permeability may lead to the enhanced cellular uptake of NPs, which in turn might result in the enhanced inhibition of HeLa cells.
To further study the cellular uptake mechanism, the intracellular distribution of FePt NPs in HeLa cells was observed qualitatively by TEM (Fig. 5B). According to TEM images, FePt NPs were absorbed and aggregated into the cells via the same endocytotic mechanism as commonly observed in other NPs systems.37,38 In this process, the NPs were engulfed in a small vesicle with invaginations of the cell membrane. Subsequently, NPs tended to be located in endosome, early lysosome, and late lysosome, gradually internalized inside the cells. In the late lysosome (pH = 5), NPs released Fe and Pt ions. Afterwards, these ions and the un-reacted FePt were freed into cytoplasm and enclosed in the nucleus, cytosol region, or released into the extracellular environment.
Considering the results demonstrated in the current study, we propose a possible mechanism accounting for FePt NPs against the HeLa cells in chemoradiotherapy as shown in Fig. 5C: after cellular uptake, under the acidic environment inside the secondary lysosomes, FePt NPs are oxidized and disintegrated to release metal ions. The released platinum ions (Pt2+) enter into the nucleus, likely bind with DNA double helix structures, interrupt the replication and transcription process, and lead to apoptosis of the HeLa cells. When combined with X-ray radiation, increased intracellular uptake of NPs might tend to release more metal ions, which in turn lead to enhanced cytotoxicity and radiosensitization effects on HeLa cells.
Overall, FePt NPs could not only act as a therapeutic agent in chemotherapy, but also perform as a radiosensitizer under the environment of radiotherapy. Hence, both the NPs concentration and the radiation dose could be reduced when NPs and radiation were combined. On the basis of this factor, it would not be farfetched to surmise that normal tissues and organs surrounding tumor cells would absorb less energy and avoid the effects of significant toxicity from FePt NP-mediated radiotherapy treatment. Coupled with the difference in the microenvironment between cancerous and normal cells, such as the permeability and the pH value, FePt nanoparticle might preferentially accumulate more in cancerous tissues than in normal tissues.14 In general, the current study proposed to utilize the interaction of FePt NPs, radiation along with the difference between the tumor and normal cells to formulate FePt NPs as improved intelligent agents in chemoradiotherapy, and thus FePt NPs may efficiently suppress the proliferation of cancer cells without inducing severe adverse effects on the normal cells. This idea could satisfy the ultimate goal of personalized chemoradiotherapy. Future work will focus on evaluating the combined effect of FePt NPs and radiation in vivo. It is noteworthy that the toxicity is the important concern for applications in clinic. The current in vitro investigations demonstrated that no noticeable cytotoxicity of FePt NPs was observed in the tested normal cells. However, considering the differences of microenvironments and condition controllabilities, it is unclear whether the current in vitro findings also apply in vivo. Thus, further research should explore the optimal nanoparticles concentration and radiation doses to increase the therapeutic ratio without causing apparent systemic toxicity.
Conclusion
The present study reported the synthesis and characterization of a family of FexPt100−x NPs with tunable compositions: Fe26Pt74, Fe53Pt47 and Fe77Pt23 NPs. After ligand exchange, the cytotoxicity of cysteamine capped FexPt100−x NPs was assessed in HEK293T cells while the contribution of Fe or Pt to the cytotoxicity and the potential application of FePt NPs in the integration of chemo- and radiotherapy were investigated in HeLa cells in vitro. It was found that FePt NPs inhibited the growth of HeLa cells in a concentration- and composition-dependent manner after 24 h incubation, with low cytotoxicity to HEK293T cells at the given concentrations. The combination of FePt NPs and radiotherapy resulted in a marked inhibition of HeLa cells, in contrast with that of the individual FePt NPs treated group or the radiation alone group. Taking the results together, FePt NPs might serve as a promising nanoprobe in chemoradiotherapy. Additionally, Fe53Pt47 NPs, exhibiting stronger cytotoxicity and enhanced radiosensitization effects on HeLa cells without damage to HEK293T cells, might theoretically improve the outcome of tumor chemoradiotherapy. Therefore, FePt NPs may in principle be functionalized with any kind of cancer-targeting antibody or peptide for further applications and improving the therapeutic efficacy. Future work will focus on further exploration of the effects of radiation time and fractionated radiotherapy on the tumor inhibition in chemoradiotherapy.Acknowledgements
This study was financially supported by the Natural Science Foundation of China (No. 10875092 and 31271511), and the Natural Science Foundation of Hubei Province of China (No. 2012KB04449). The authors are grateful to the Department of Radio- and Chemo-therapy, Zhongnan Hospital of Wuhan University, Wuhan, China, and Elekta Instrument (Shanghai) Ltd. for their assistance with the project.References
- A. Jemal, F. Bray, M. M. Center, J. Ferlay, E. Ward and D. Forman, Ca-Cancer J. Clin., 2011, 61, 69–90 CrossRef PubMed.
- J. Ferlay, H. R. Shin, F. Bray, D. Forman, C. Mathers and D. M. Parkin, Int. J. Cancer, 2010, 127, 2893–2917 CrossRef CAS PubMed.
- Z. H. Siddik, Oncogene, 2003, 22, 7265–7279 CrossRef CAS PubMed.
- R. Sinha, G. J. Kim, S. Nie and D. M. Shin, Mol. Cancer Ther., 2006, 5, 1909–1917 CrossRef CAS PubMed.
- T. M. Stortecky and S. Suter, Curr. Opin. Oncol., 2010, 22, 312–317 CrossRef PubMed.
- A. Fujiwara-Igarashi, T. Fujimori, M. Oka, Y. Nishimura, Y. Hamamoto, Y. Kazato, H. Sawada, N. Yayoshi, D. Hasegawa and M. Fujita, Vet. J., 2014, 202, 455–461 CrossRef PubMed.
- A. Magdalena, M. Panayiotis, L. K. Bengt and B. Anders, Cancers, 2011, 3, 2421–2443 CrossRef PubMed.
- C. L. Emory, C. O. Montgomery, B. K. Potter, M. E. Keisch and S. A. Conway, Clin. Orthop. Relat. Res., 2012, 470, 751–758 CrossRef PubMed.
- N. P. Rowell and N. O'Rourke, Cochrane Database of Systematic Reviews, 2010, 65, D2140 Search PubMed.
- M. Kawashima, Int. J. Clin. Oncol., 2004, 9, 421–434 CrossRef PubMed.
- D. Brandsma, L. Stalpers, W. Taal, P. Sminia and M. J. V. D. Bent, Lancet Oncol., 2008, 9, 453–461 CrossRef PubMed.
- J. H. Park, E. K. Choi, S. D. Ahn, S. W. Lee, Y. S. Si, M. Y. Sang, Y. S. Kim, S. L. Yu, S. G. Lee and S. Hwang, Int. J. Radiat. Oncol., Biol., Phys., 2011, 79, 696–704 CrossRef CAS PubMed.
- G. Yu, J. Xie, H. Chen, S. Gu, R. Zhao, J. Shao and L. Jia, Biotechnol. Adv., 2014, 32, 761–777 CrossRef PubMed.
- J. D. Rocca, M. E. Werner, S. A. Kramer, R. C. Huxford-Phillips, R. Sukumar, N. D. Cummings, J. L. Vivero-Escoto, A. Z. Wang and W. Lin, Nanomedicine, 2015, 11, 31–38 CAS.
- E. C. Wang, M. Yuanzeng, R. C. Palm, J. J. Fiordalisi, K. T. Wagner, H. Nabeel, A. D. Cox, J. M. Caster, T. Xi and A. Z. Wang, Biomaterials, 2015, 51, 208–215 CrossRef CAS PubMed.
- S. Tianmeng, Z. Yu Shrike, P. Bo, H. D. Choon, Y. Miaoxin and X. Younan, Angew. Chem., Int. Ed., 2014, 53, 12320–12364 Search PubMed.
- H. Maeda, Bioconjugate Chem., 2010, 21, 797–802 CrossRef CAS PubMed.
- J. W. J. Bergs, M. G. Wacker, S. Hehlgans, A. Piiper, G. Multhoff, C. R. Del and F. R. Del, Biochim. Biophys. Acta, Rev. Cancer, 2015, 1856, 130–143 CrossRef CAS PubMed.
- J. U. Menon, V. Tumati, J. T. Hsieh, K. T. Nguyen and D. Saha, J. Biomed. Mater. Res., Part A, 2015, 103, 1632–1639 CrossRef PubMed.
- H. Zahra, K. Samideh, K. Sepideh and M. Seied Rabi, IEEE Transactions on NanoBioscience, 2014, 13, 403–408 CrossRef PubMed.
- C. Jin, L. Bai, H. Wu, J. Liu, G. Guo and J. Chen, Cancer Biol. Ther., 2008, 7, 911–916 CrossRef CAS PubMed.
- G. Sebastian, R. Sebastian, R. D. Claus, R. D. Franz and K. J. Rg, J. Microencapsulation, 2012, 29, 685–694 CrossRef PubMed.
- C. Jing, Z. Shu, L. Tong, J. Li, C. Fei, Y. Han, Z. Ming and X. Wei, BMC Cancer, 2014, 14, 1–8 CrossRef PubMed.
- F. B. Cui, Q. Liu, R. T. Li, J. Shen, P. Y. Wu, L. X. Yu, W. J. Hu, F. L. Wu, C. P. Jiang and G. F. Yue, Int. J. Nanomed., 2014, 9, 2345–2358 Search PubMed.
- X. Zhang, D. Wu, X. Shen, J. Chen, Y. Sun, P. Liu and X. Liang, Biomaterials, 2012, 33, 6408–6419 CrossRef CAS PubMed.
- P. Liu, Z. Huang, Z. Chen, R. Xu, H. Wu, F. Zang, C. Wang and N. Gu, Nanoscale, 2013, 5, 11829 RSC.
- W. N. Rahman, N. Bishara, T. Ackerly, C. F. He, P. Jackson, C. Wong, R. Davidson and M. Geso, Nanomedicine, 2009, 5, 136–142 CAS.
- S. Sun, Adv. Mater., 2006, 18, 393–403 CrossRef CAS.
- H. Rui, X. Ruijun, X. Zhichuan, H. Yanglong, G. Song and S. Shouheng, Adv. Mater., 2010, 22, 2729–2742 CrossRef PubMed.
- S. Chou, Y. Shau, P. Wu, Y. Yang, D. Shieh and C. Chen, J. Am. Chem. Soc., 2010, 132, 13270–13278 CrossRef CAS PubMed.
- J. Gao, G. Liang, B. Zhang, Y. Kuang, X. Zhang and B. Xu, J. Am. Chem. Soc., 2007, 129, 1428–1433 CrossRef CAS PubMed.
- J. Gao, G. Liang, J. S. Cheung, Y. Pan, Y. Kuang, F. Zhao, B. Zhang, X. Zhang, E. X. Wu and B. Xu, J. Am. Chem. Soc., 2008, 130, 11828–11833 CrossRef CAS PubMed.
- S. Chen, L. Wang, S. L. Duce, S. Brown, S. Lee, A. Melzer, S. A. Cuschieri and P. André, J. Am. Chem. Soc., 2010, 132, 15022–15029 CrossRef CAS PubMed.
- Y. Zheng, Y. Tang, Z. Bao, H. Wang, F. Ren, M. Guo, H. Quan and C. Jiang, Int. J. Nanomed., 2015, 10, 6435–6444 CrossRef PubMed.
- V. Nandwana, K. E. Elkins, N. Poudyal, G. S. Chaubey, K. Yano and J. P. Liu, J. Phys. Chem. C, 2007, 111, 4185–4189 CAS.
- W. Qingzhi, C. Huaqiang, L. Qiuying, Z. Jiyong, W. Zhao, J. H. Warner and A. A. R. Watt, Inorg. Chem., 2008, 47, 5882–5888 CrossRef PubMed.
- Y. Fan, C. Li, H. Cao, F. Li and D. Chen, Biomaterials, 2012, 33, 4220–4228 CrossRef CAS PubMed.
- J. Park, H. Kim, J. Kim, S. Jeong, J. Jung, G. Lee, J. Lee, Y. Chang and T. Kim, Bioorg. Med. Chem. Lett., 2010, 20, 2287–2291 CrossRef CAS PubMed.
- K. Li, D. Ding, D. Huo, K. Pu, N. N. P. Thao, Y. Hu, Z. Li and B. Liu, Adv. Funct. Mater., 2012, 22, 3107–3115 CrossRef CAS.
- H. K. Patra, S. Banerjee, U. Chaudhuri, P. Lahiri and A. K. Dasgupta, Nanomedicine, 2007, 3, 111–119 CAS.
- W. Gao, L. Ji, L. Li, G. Cui, K. Xu, P. Li and B. Tang, Biomaterials, 2012, 33, 3710–3718 CrossRef CAS PubMed.
- S. Ostrovsky, G. Kazimirsky, A. Gedanken and C. Brodie, Nano Res., 2009, 2, 882–890 CrossRef CAS.
- S. Jain, J. A. Coulter, A. R. Hounsell, K. T. Butterworth, S. J. McMahon, W. B. Hyland, M. F. Muir, G. R. Dickson, K. M. Prise, F. J. Currell, J. M. O' Sullivan and D. G. Hirst, Int. J. Radiat. Oncol., Biol., Phys., 2011, 79, 531–539 CrossRef CAS PubMed.
- J. Shaw, S. O. Raja and A. K. Dasgupta, Cancer Nanotechnol., 2014, 5, 1–15 CrossRef PubMed.
- M. J. Akhtar, H. A. Alhadlaq, S. Kumar, S. A. Alrokayan and M. Ahamed, Arch. Toxicol., 2015, 89, 1895–1907 CrossRef CAS PubMed.
- M. Moglianetti, E. De Luca, D. Pedone, R. Marotta, T. Catelani, B. Sartori, H. Amenitsch, S. F. Retta and P. P. Pompa, Nanoscale, 2016, 8, 3739–3752 RSC.
- H. Sun, X. Chen, C. Dan, M. Dong, X. Fu, L. Qian, L. Xi, Q. Wu, Q. Tong and W. Tao, Int. J. Nanomed., 2012, 7, 3295–3307 CAS.
- S. Tianmeng, Z. Yu Shrike, P. Bo, H. D. Choon, Y. Miaoxin and X. Younan, Angew. Chem., Int. Ed., 2014, 53, 12320–12364 Search PubMed.
- M. M. Gottesman, T. Fojo and S. E. Bates, Nat. Rev. Cancer, 2002, 2, 48–58 CrossRef CAS PubMed.
- K. Cho, X. Wang, S. Nie, Z. Chen and D. M. Shin, Clin. Cancer Res., 2008, 14, 1310–1316 CrossRef CAS PubMed.
- T. Nakazawa and S. Nagatsuka, Int. J. Radiat. Biol. Relat. Stud. Phys., Chem. Med., 2009, 38, 537–544 CrossRef.
- C. Guozhen, Z. Miaomiao, M. Jianshun, L. Wenjian, W. Jufang, L. Dong and X. Jiefang, J. Radiat. Res., 2015, 56, 294–304 CrossRef PubMed.
Footnote |
| † Z. R. Bao and M. Y. He contributed equally to the manuscript. |
| This journal is © The Royal Society of Chemistry 2016 |