
Dynamic polyrotaxane-coated surface for effective differentiation of mouse induced pluripotent stem cells into cardiomyocytes
Abstract
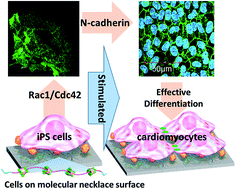
The effect of increasing molecular mobility, on hydrated polyrotaxane (PRX)-coated surfaces, on differentiation of mouse induced pluripotent stem cells (iPS cells) into cardiomyocytes was examined. PRX is composed of α-cyclodextrin (α-CD) threaded on linear poly(ethylene glycol) (PEG)-capped terminals with bulky end-groups. The degree of molecular mobility at the hydrated state (Mf) on the PRX surfaces can be varied by changing the number of threaded α-CDs. Rac1 expression was significantly upregulated for adhering iPS cells on the PRX surface with high Mf value, while it was downregulated on surfaces with low Mf value. Furthermore, the expression of N-cadherin, which is an important marker protein for cardiomyogenic differentiation of stem cells, was greatly upregulated for adhering iPS cells on the PRX surface with high Mf value, while those on surfaces with low Mf value showed low N-cadherin expression. Finally, the PRX surface with higher Mf value was found to be higher in cardiomyogenesis and beating colony formation from iPS cells, the extent of which was much higher than that on gelatin-coated surfaces. This suggests that surface hydrated molecular mobility, varied by varying a supramolecular PRX architecture on materials, plays a significant role in controlling cytoskeletal signaling pathways, eventually contributing to the direction of stem cell commitment.
 Please wait while we load your content...
Please wait while we load your content...

