
In situ synthesis of carbon fiber-supported SiOx as anode materials for lithium ion batteries†
Xuejun Baib,
Biao Wang*ab,
Huaping Wangab and
Jianming Jiangb
aCollege of Material Science and Engineering, Donghua University, Shanghai, P. R. China 201620. E-mail: wbiao2000@dhu.edu.cn
bState Key Laboratory for Modification of Chemical Fibers and Polymer Materials, Donghua University, Shanghai, P. R. China 201620
First published on 24th March 2016
Abstract
The carbon fiber-supported SiOx (CF–SiOx) composites are in situ fabricated using a facile two-step method and evaluated as anodes for lithium-ion batteries (LIBs). The CF–SiOx anodes exhibit a reversible capacity of 1100 mA h g−1 at 50 mA g−1 after a high current charge–discharge cycling test with a capacity retention of 80% (compared with the first 5 cycles). This superior performance of the CF–SiOx are due to the modification of SiOx with the profiled carbon fibers, which effectively construct a continuous conducting network and thus enhance the electrochemical activity of SiOx. The profiled carbon fibers have numerous surface grooves to provide high contact area for rapid lithium-ion transport and enough inter-fiber space for the accommodation of large SiOx volume changes on lithium insertion and extraction.
1 Introduction
Lithium-ion batteries (LIBs) have become the dominant power sources for portable electronic devices and important power for vehicles and aerospace due to their long cycle life, safety, high energy and power densities.1–5 Based on these various applications, there is growing demand for next generation LIBs with high energy and power density.6–8 The low theoretical gravimetric capacity of graphite (372 mA h g−1), a commercial anode material, can't satisfy the demands.9,10 So, researchers have devoted significant attentions to develop high capacity anode electrode materials for LIBs. Among them, silicon has attracted considerable interest due to its abundant sources, low cost, high theoretical capacities (∼4200 mA h g−1), and relatively low working potential (∼0.5 V vs. Li/Li+).11,12 However, the electrochemical alloying reactions of Si with Li involve volumetric expansion of up to 400% during lithium insertion.13,14 This stress, induced by the large volume change, causes cracking and pulverization of silicon, which leads to the loss of electrical contact and early capacity fading of the battery.15,16In recent years, more attention has been paid to Silicon oxide anode materials such as SiO, SiOx and SiO2.17–20 These Silicon oxides react with Li to form Si, LixSi and irreversible Li4SiO4, Li2O during the initial lithiathion process.19 The irreversible products can act as a buffer component to improve the cycling performance of electrodes.21,22 The theoretical specific capacity of SiO2 is calculated to be 1965 mA h g−1, but such high capacity is often difficult to achieve for the reason of volumetric expansion effect, low electrical conductivity, and generation of irreversible lithium silicate.23 Many trials have been devoted to coping with this problem such as optimizing the morphology,18,22,24,25 decreasing the size,17,19,26 adding the electrodes with buffer materials,27–29 and coating of active materials with electrically conducting materials.30–32 Among these methods, the electrochemical performance of SiO anodes can be considerably improved by adding electrical conductive buffer materials. However the volume expansion, agglomeration, and detachment of SiO can not be totally avoided. On the other hand, the synthetic methods of these materials are too complicate. Therefore, researches should be focused on the prevention of agglomeration of the derived nanoscale Si and finding a facile synthetic method for well-designed SiOx-based composites towards high-performance anode materials.
In this work, the novel architecture of profiled carbon fiber-supported SiOx composites is developed. The profiled carbon fibers with numerous surface grooves provide high contact area for CF–SiOx composites with electrolyte to increase reaction extent with Li+, and enough inter-fiber space for the accommodation of large SiOx volume changes on lithium insertion and extraction. In addition, this carbon fibers can also form a electrical conductive network for the SiOx particles to maintain the electrical conduction pathways of the electrodes. The novel designed profiled carbon fiber-SiOx anodes exhibit improved electrochemical properties.
2 Experimental
2.1 Sample preparation
To obtain the CF–SiOx composites, two processes were needed: coating and heat treatment. Coating process: viscose fibers (VF) and tetraethyl orthosilicate (TEOS) were served as carbon source and silicon source, respectively. VF was pre-treated in 1 M HCl aqueous solution for 1 h to remove the impurities, followed by washing and drying. 9 g of pre-treated VF was impregnated in 300 mL NH4OH (Sinopharm Chemical Reagent Co., Ltd) ethanol solution for 20 h, followed by adding 200 mL TEOS (AR, Aladdin) ethanol solution drop-wise. The reaction mixtures were allowed to proceed for 24 h under vigorous stirring, after which excess liquid was removed with filter papers and dried at 60 °C overnight. Table 1 shows the amount of reagents employed for the preparation of different composite samples. Heat treatment process: the impregnated viscose fibers were placed in a gas flow reactor consisting of a quartz glass tube and a furnace fitted with an independent temperature controller. A 10 cm3 min−1 flow of Ar/H2 (H2 15 vol%) was introduced into the quartz tube during heating process. The temperature was slowly increased to 400 °C at a rate of 2 °C min−1 and maintained for 1 h. Then temperature was continuously increased to 800 °C at a rate of 5 °C min−1 and maintained for 2 h.| Sample | VF (g) | NH4OH (mol L−1) | TEOS (mol L−1) |
|---|---|---|---|
| CF–SiOx-1 | 9 | 0.15 | 0.7 |
| CF–SiOx-2 | 9 | 0.17 | 0.75 |
| CF–SiOx-3 | 9 | 0.2 | 0.9 |
| CF–SiOx-4 | 9 | 0.4 | 1.8 |
| SiOx | 0 | 0.2 | 0.9 |
For comparison, both pure SiOx particles and carbon fibers (CF) were prepared. SiOx particles: 200 mL TEOS ethanol solution was added in 300 mL NH4OH ethanol solution drop-wise. The reaction solution was allowed to proceed for 24 h under vigorous stirring. The white sediments (SiOx particles) were repeatedly washed with ethanol and separated using high-speed centrifugation (20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 1 h) until the pH of the solution became neutral. Then SiOx particles were vacuum-dried at 80 °C overnight, followed by heat treatment mentioned above. The preparation of the carbon fibers (CF) has already reported in our previous work.3
000 rpm for 1 h) until the pH of the solution became neutral. Then SiOx particles were vacuum-dried at 80 °C overnight, followed by heat treatment mentioned above. The preparation of the carbon fibers (CF) has already reported in our previous work.3
2.2 Sample characterization
X-ray diffraction spectroscopy (XRD) was carried out by a Rigaku D/max 2550 V X-ray diffractometer using Cu-Kα source. Raman spectrum was measured on a T64000 triple Raman system using the 514.5 nm Ar-ion laser as an excitation source. X-ray photoelectron spectroscopy (XPS) was conducted by Thermo Scientific ESCALAB 250Xi with X-ray source of Al Kα (1486.6 eV). Thermogravimetric analysis (TGA) was recorded on a Netzsh TG209 F1analyzer, from room temperature to 800 °C with a ramp rate of 15 °C min−1 and in air (15 mL min−1). High-resolution field emission scanning electron microscopy (FESEM, Hitachi S-4800) was used to characterized the morphologies of samples. Cycled electrodes were rinsed in dimethyl carbonate (DMC) in an argon-filled glovebox before characterized by FESEM.2.3 Electrochemical measurements
The electrochemical performances were determined by assembling two-electrode coin-type (CR2016) half cells with Celgard 2400 as the separator and lithium foil as counter electrode. The electrolyte was 1 M LiPF6 in ethylene carbonate/dimethyl carbonate (EC/DMC 1:1 v/v). The working electrodes were prepared by coating a mixture of 80 wt% CF–SiOx materials, 10 wt% conductive material (acetylene black) and 10 wt% binder (polyvinylidene fluoride) onto a copper foil current collector, then vacuum-dried at 80 °C for 12 h. Electrode loading of CF–SiOx composites is 1.8 mg. The coin cells were assembled in an argon-filled glove box where oxygen and water concentration were strictly limited below 1 ppm. The galvanostatic cycling measurements were performed on CT2001A LAND battery testing system in the voltage range of 0.01–2 V vs. Li+/Li. The current setting for cell tests and the specific capacity were calculated based on the mass of CF–SiOx materials on working electrodes. Cyclic voltammogram (CV) and electrochemical impedance spectroscopy (IES) were recorded by electrochemical workstation (Autolab PGSTAT128N). CV was tested at a scan rate of 0.1 mV s−1 within 2–0.01 V and EIS was tested under the following conditions: open potential of 0.5 V, AC voltage amplitude of 10 mV, and frequency range from 106 to 0.01 Hz.
3 Results and discussion
Fig. 1 illustrates the preparation process of CF–SiOx composites. VF was served as the carbon source, which turned to CF during heat treatment at a high temperature of 800 °C under argon atmosphere. SiOx particles as the active anode material for LIBs were introduced around the CF. The detailed synthesis processes have been described in the experiment section. This synthesis method is very simple.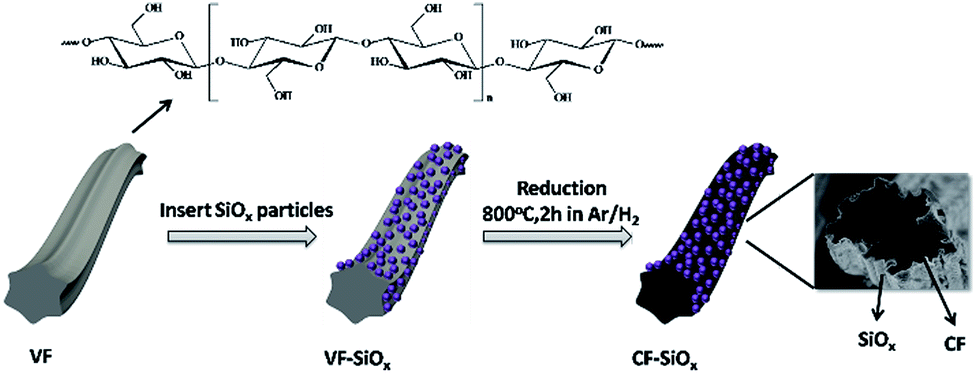 | ||
| Fig. 1 Schematic illustration of procedure to synthesize CF–SiOx composites, with a insert showing the cross-section SEM image. | ||
It is reported that the crystalline SiOx as the anode materials of LIBs can't react with Li+ due to its structural stability. However it exhibits Li+ reactivity in amorphous states.19 X-ray diffraction patterns of CF and CF–SiOx-3 composites are compared in Fig. 2a. These two patterns are almost same and no crystalline peaks of silicon oxide can be observed in CF–SiOx composites. Therefore, the SiOx synthesized by this method is amorphous and could have lithium ion storage ability. Fig. 2b shows Raman spectroscopy of CF–SiOx-3 composites. The two strong peaks at 1358 cm−1 and 1590 cm−1 are assigned to a disorder band (D band) and a strong tangential mode band (G band). ID/IG < 1 reveals that CF is partially graphitized, which could provide high electrical conductivity for CF–SiOx composites.33 The broad peak at approximately 400–500 cm−1 demonstrates the formation of amorphous SiOx.21 In order to confirm the form of C and the valence state of the Si in CF–SiOx composites, X-ray photoelectron spectroscopy (XPS) has been conducted (Fig. 2c–e). The C1s spectrum of CF–SiOx-3 yields to two peaks: a weak peak at 286.7 eV and a main peak at 284.7 eV, which present the carbon in the O–C–O/C–O and C/C moieties, respectively. The peak at 284.7 eV, which is associated to sp2 C, is much stronger and becomes a dominant peak. The peak at 286.7 eV is very weak, suggesting the carbonized degree of carbon fibers is very high and the organic groups are almost disappeared. This may provide excellent conductivity for these materials used in LIBs. XPS high resolution Si 2p spectrum (Fig. 2e) clearly displays that three valence states of Si exist in these samples which are Si4+ (103.8 eV), Si3+ (103 eV) and Si2+ (101.8 eV). Si4+ is the major ingredient (87%) and the peaks of Si3+ and Si2+ are very weak which show their contents are pretty low (6.4% and 6.5% respectively). And the O/Si atomic ratio in XPS results is almost 2, indicating that SiO2 is the main component in the composites. Though Si3+ and Si2+ are not stable, their activities with Li+ are a little higher than that of Si4+ in LIBs. And their existences are expected to be benefit to increase the capacity and rate charge/discharge ability of CF–SiOx composites. The SiOx contents in these composites are determined using thermogravimetric analysis (TGA) and the results are shown in Fig. 2f. The SiOx contents in samples CF–SiOx-1, -2, -3, and -4 are 4.3 wt%, 9.8 wt%, 18.5 wt% and 23.7 wt% respectively, by the remaining weight after heating to above 700 °C. The contents of SiOx increase with the concentration of NH4OH and TEOS, when the ratio of these two reactants is constant (see Table 1). The concentrations of reactants also have obvious effects on the microstructure of CF–SiOx composites.34,35
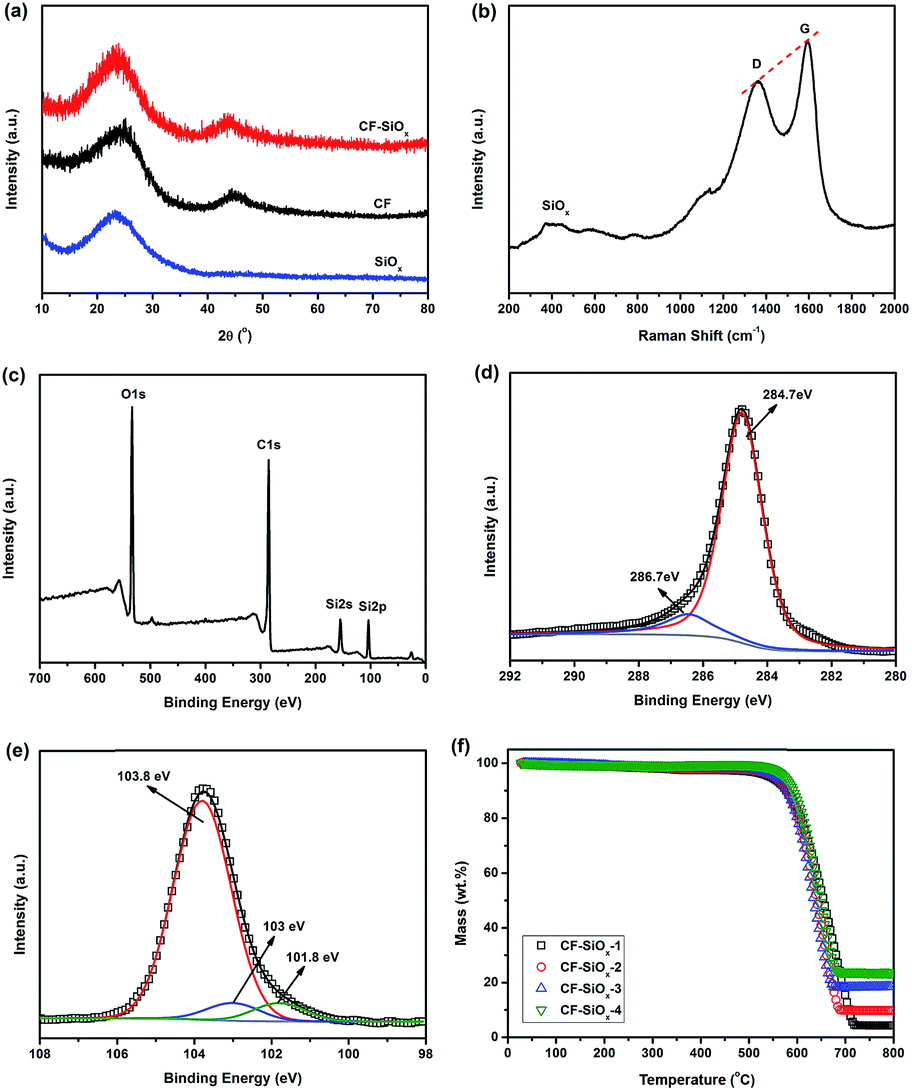 | ||
| Fig. 2 (a) XRD patterns of CF, CF–SiOx-3, and SiOx; (b) Raman spectrum for CF–SiOx-3; (c) XPS spectra of survey scan, (d) high resolution C 1s and (e) Si 2p for CF–SiOx-3; (f) thermogravimetric analysis curves of the CF–SiOx composites. | ||
The FESEM images of CF–SiOx composites are shown in Fig. 3. As shown in Fig. S1,† the CF exhibits uneven surface and a profiled cross-section which could provide large surface area and multiple inter-fiber spaces. After the sol–gel reaction and carbonization process, SiOx particles show homogenous distribution on the surface of CF and cross-section of CF–SiOx composites are similar to CF (Fig. 3). But the size of SiOx particles is so different (Fig. 3 insets), which is really affected by the concentration of TEOS to NH4OH as mentioned above. When the diameter of SiOx particles is smaller than 5 nm (sample CF–SiOx-1, -2), these particles agglomerate together heavily and form the corrugation. If the diameters increase to 20 nm or 50 nm (sample CF–SiOx-3, -4), the SiOx particles distribute independently and loose stack on the CF surface. For the pure SiOx particles, although the diameter is almost the same with that of CF–SiOx-3 (Fig. S2†), they are not monodisperse particles and are agglomerated. This suggests that the utilization of VF can considerably improve uniformity of particles dispersion. The size and aggregation morphology of SiOx particles may have effects on electrochemistry performances.
 | ||
| Fig. 3 FESEM images of various CF–SiOx composites, (a, b) CF–SiOx-1, (c, d) CF–SiOx-2, (e, f) CF–SiOx-3, (g, h) CF–SiOx-4; (a, c, e, g) are top surface images, with insets showing the images with higher magnification; (b, d, f, h) are cross-section images, with a inset showing the EDS mapping of Si (yellow). | ||
Fig. 4a shows the cyclic voltammogram (CV) profiles of the CF–SiOx-3 composites. In the cathodic polarization process of the 1st cycle, the cathodic peat at 0.75 V can be ascribed to the formation of SEI layers on the surface of SiOx particles and CF.21 The cathodic peak at 0.35 V is attributed to the reduction of SiOx to Si and the formation of Li2O and LiSiO4.19 The cathodic peak at about 0.02 V corresponds to the alloying of Si with Li and the reversible reaction between Li and CF. In the anodic polarization process, the peak at 0.15 V is related to the de-alloying of the Li–Si alloys. After the 3rd cycle, the near overlap of the curves implies good capacity retention. Fig. 4b shows the cycling performances of CF–SiOx composites at 250 mA g−1 after 10 cycles at 50 mA g−1 when batteries are much more stable. These CF–SiOx composites deliver reversible capacities of 400, 473, 614 and 665 mA h g−1 in the 1st cycle (at 250 mA g−1) and then remain 243, 284, 481 and 285 mA h g−1 after 150 cycles. The capacity retentions are 61%, 60%, 78%, 43%, respectively. For comparison, CF and pure SiOx particles anodes are synthesized and tested. CF show stable capacities of 200 mA h g−1 and SiOx particles exhibit a reversible capacity of 1397 mA h g−1 for the 1st cycle and then the capacity drops below 100 mA h g−1 after 50 cycles (Fig. S3†). These results suggest that effective availability of SiOx in these composites considerably increases due to the conductive network constructed by CF. Both SiOx contents and the particle size have effects on the cycling performance. The initial capacity increases with the SiOx contents, but the cycling stability shows different trend. For CF–SiOx-1, -2, the SiOx nanoparticles agglomerate together very heavily, leading to 40% loss of capacity after 150 cycles test. As mentioned before (Fig. 3), with the increases of SiOx contents and particle size, the particles are monodispersed. CF–SiOx-3, the loose stacking of SiOx particles provides void space for Li diffusion channels and allows electrolytes to penetrate easily into the CF and react electrochemically with SiOx. Moreover, the uniform distribution of SiOx on CF surface and multiple inter-fiber spaces can effectively accommodate the volumetric variation during cycling and consequently leading to the long-term cycling stability. After 150 cycles, there is no visible damage to the initial structure of CF–SiOx-3 (Fig. 4c). But when the size of SiOx increases to upper 50 nm (CF–SiOx-4) in size, the absolute volumetric change is twice as big as the CF–SiOx-3. This causes SiOx severe agglomeration and shedding from CF during charge–discharge process, leading to a rapid fading of capacity. The galvanostatic voltage profiles of CF–SiOx-3 at selected cycles are also shown in Fig. S4.†
 | ||
| Fig. 4 (a) Cyclic voltammograms of the CF–SiOx-3 composites in the initial ten cycles; (b) cycling performances of CF–SiOx composites at 250 mA g−1 between 0.01 and 2.0 V; (c) SEM images of CF–SiOx-3 anode after 150 cycles. | ||
The rate capability of the CF–SiOx-3 up to 500 mA g−1 is shown in Fig. 5. The average capacities at these rates are 1340, 1200, 850 and 370 mA h g−1 for 50, 100, 200 and 500 mA g−1, respectively. These outstanding capacities well prove the rate capability offered by this profiled fiber structure. When the current rate is back to 50 mA g−1, the capacity recovers to 1100 mA h g−1, indicating capacity retention of 80%.
 | ||
| Fig. 5 Specific capacities of CF–SiOx-3 at various current densities as marked. The voltage is between 0.01 and 2.0 V. | ||
The Nyquist plots obtained at the 3rd cycles for CF–SiOx composites are used to evaluate the resistance against electron transfer during cycling (Fig. 6). The electrochemical plots are fitted by an equivalent circuit (Fig. 6 inset). The semicircle in the high-frequency region is associated with charge transfer impedance (Rct) and constant phase element of the electrode/electrolyte interface (CPE). The line in the low-frequency region is the Warburg impedance (Wo), which is related to the diffusion of lithium ions into the bulk of the electrode material. Table 2 shows the parameters of the equivalent circuit for all composites after fitting. The Rs of these composites is relatively low, which indicates that electrical conductivity of these samples is good and the SEI films are stable. But the Rct of these samples is quite different from each other. As mentioned above, the SiOx particles of CF–SiOx-1, -2 are packed closely around CF and the SiOx shell of CF–SiOx-3, -4 is much looser, which provides abundant lithium-ion transfer channels. This enhances the electrochemical activity and lithium-ion rate storage performance of these composites anode material.
 | ||
| Fig. 6 The EIS of CF–SiOx composites after 3 cycles, with inset shows the equivalent circuit. | ||
| Sample | Rs (Ω) | CPE-T | CPE-P | Rct (Ω) |
|---|---|---|---|---|
| SiOx/CF-1 | 3.3 | 2.9 × 10−5 | 0.8 | 114.2 |
| SiOx/CF-2 | 2.8 | 6.8 × 10−5 | 0.6 | 144.3 |
| SiOx/CF-3 | 3.1 | 3.5 × 10−5 | 0.7 | 46.9 |
| SiOx/CF-4 | 3.4 | 2.6 × 10−5 | 0.7 | 59.1 |
4 Conclusions
CF–SiOx composites as anode materials for LIBs are in situ fabricated via a simple two-step process. The size and morphology of loaded SiOx particles can be controlled by the concentration of reactants. The profiled carbon fibers provide large surface area and enough inter-fiber space for efficient loading of SiOx, accommodating volume charges of SiOx, abundant lithium-ion transfer channels and good penetration of electrolyte solutions. The CF–SiOx composites produce stable capacity of 1100 mA h g−1 at 50 mA g−1, after high current charge–discharge cycling test. Even at 250 mA g−1, the electrodes exhibit a high reversible capacity of 481 mA h g−1 for 150 cycles with a 78% capacity retention, which reveals a high Li-ion diffusion rate of the electrodes. The simple and applicable synthesis process also provides a promising method for preparing carbon fiber-based anode materials for high performance LIBs.Acknowledgements
This work was financially supported by the Shanghai Leading Academic Discipline Project (No. B603) and the Program of Introducing Talents of Discipline to Universities (No. 111-2-04). Xuejun Bai was also supported by a fellowship from the Chinese Scholarship Council. Assistance from the research groups of Prof. Harold H. Kung and Prof. Samuel Stupp of Northwestern University for the XPS spectra and EIS tests was acknowledged.Notes and references
- B. Dunn, H. Kamath and J. M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS PubMed.
- S. Q. Chen, P. T. Bao, X. D. Huang, B. Sun and G. X. Wang, Nano Res., 2014, 7, 85–94 CrossRef CAS.
- X. J. Bai, B. Wang, H. P. Wang and J. M. Jiang, J. Alloys Compd., 2015, 628, 407–412 CrossRef CAS.
- Y. Li, J. Song and J. Yang, Renewable Sustainable Energy Rev., 2014, 37, 627–633 CrossRef.
- T. H. Kim, J. S. Park, S. K. Chang, S. Choi, J. H. Ryu and H. K. Song, Adv. Energy Mater., 2012, 2, 860–872 CrossRef CAS.
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed.
- J. B. Goodenough and Y. Kim, Chem. Mater., 2010, 22, 587–603 CrossRef CAS.
- O. K. Park, Y. Cho, S. Lee, H. C. Yoo, H. K. Song and J. Cho, Energy Environ. Sci., 2011, 4, 1621–1633 CAS.
- W. Li, Y. Tang, W. Kang, Z. Zhang, X. Yang, Y. Zhu, W. Zhang and C. S. Lee, Small, 2015, 11, 1345–1351 CrossRef CAS PubMed.
- H. Wu, G. Zheng, N. Liu, T. J. Carney, Y. Yang and Y. Cui, Nano Lett., 2012, 12, 904–909 CrossRef CAS PubMed.
- Q. Si, K. Hanai, T. Ichikawa, A. Hirano, N. Imanishi, O. Yamamoto and Y. Takeda, J. Power Sources, 2011, 196, 6982–6986 CrossRef CAS.
- J. B. Chang, X. K. Huang, G. H. Zhou, S. M. Cui, P. B. Hallac, J. W. Jiang, P. T. Hurley and J. H. Chen, Adv. Mater., 2014, 26, 758–764 CrossRef CAS PubMed.
- Q. Si, K. Hanai, T. Ichikawa, A. Hirano, N. Imanishi, Y. Takeda and O. Yamamoto, J. Power Sources, 2010, 195, 1720–1725 CrossRef CAS.
- M. H. Park, M. G. Kim, J. Joo, K. Kim, J. Kim, S. Ahn, Y. Cui and J. Cho, Nano Lett., 2009, 9, 3844–3847 CrossRef CAS PubMed.
- M. T. McDowell, S. W. Lee, I. Ryu, H. Wu, W. D. Nix, J. W. Choi and Y. Cui, Nano Lett., 2011, 11, 4018–4025 CrossRef CAS PubMed.
- C. D. Wang, Y. Li, Y. S. Chui, Q. H. Wu, X. F. Chen and W. J. Zhang, Nanoscale, 2013, 5, 10599–10604 RSC.
- B. K. Guo, J. Shu, Z. X. Wang, H. Yang, L. H. Shi, Y. N. Liu and L. Q. Chen, Electrochem. Commun., 2008, 10, 1876–1878 CrossRef CAS.
- Q. Sun, B. Zhang and Z. W. Fu, Appl. Surf. Sci., 2008, 254, 3774–3779 CrossRef CAS.
- W. S. Chang, C. M. Park, J. H. Kim, Y. U. Kim, G. Jeong and H. J. Sohn, Energy Environ. Sci., 2012, 5, 6895–6899 CAS.
- B. C. Yu, Y. Hwa, J. H. Kim and H. J. Sohn, Electrochim. Acta, 2014, 117, 426–430 CrossRef CAS.
- C. F. Guo, D. L. Wang, T. F. Liu, J. S. Zhu and X. S. Lang, J. Mater. Chem. A, 2014, 2, 3521–3527 CAS.
- Z. Favors, W. Wang, H. H. Bay, A. George, M. Ozkan and C. S. Ozkan, Sci. Rep., 2014, 4, 4605–4612 Search PubMed.
- N. Yan, F. Wang, H. Zhong, Y. Li, Y. Wang, L. Hu and Q. W. Chen, Sci. Rep., 2013, 3, 1568–1574 Search PubMed.
- M. Sasidharan, D. Liu, N. Gunawardhana, M. Yoshio and K. Nakashima, J. Mater. Chem., 2011, 21, 13881–13888 RSC.
- D. Qiu, G. Bu, B. Zhao and Z. Lin, J. Solid State Electrochem., 2015, 19, 935–939 CrossRef CAS.
- J. Xie, G. Wang, Y. Huo, S. Zhang, G. Cao and X. Zhao, Electrochim. Acta, 2014, 135, 94–100 CrossRef CAS.
- J. Wang, H. Zhao, J. He and C. Wang, J. Power Sources, 2011, 196, 4811–4815 CrossRef CAS.
- S. B. Yang, X. L. Feng, L. Wang, K. Tang, J. Maier and K. Mullen, Angew. Chem., Int. Ed., 2010, 49, 4795–4799 CrossRef CAS PubMed.
- D. T. Nguyen, C. C. Nguyen, J. S. Kim, J. Y. Kim and S. W. Song, ACS Appl. Mater. Interfaces, 2013, 5, 11234–11239 CAS.
- L. Su, Z. Zhou and M. Ren, Chem. Commun., 2010, 46, 2590–2592 RSC.
- S. B. Yang, X. L. Feng, S. Ivanovici and K. Mullen, Angew. Chem., Int. Ed., 2010, 49, 8408–8411 CrossRef CAS PubMed.
- D. J. Lee, M. H. Ryou, J. N. Lee, B. G. Kim, Y. M. Lee, H. W. Kim, B. S. Kong, J. K. Park and J. W. Choi, Electrochem. Commun., 2013, 34, 98–101 CrossRef CAS.
- R. Chen, T. Zhao, J. Lu, F. Wu, L. Li, J. Chen, G. Tan, Y. Ye and K. Amine, Nano Lett., 2013, 13, 4642–4649 CrossRef CAS PubMed.
- J. W. Kim, L. U. Kim and C. K. Kim, Biomacromolecules, 2007, 8, 215–222 CrossRef CAS PubMed.
- W. Stöber, A. Fink and E. Bohn, J. Colloid Interface Sci., 1968, 26, 62–69 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra03963d |
| This journal is © The Royal Society of Chemistry 2016 |