
Controlled nanogel and macrogel structures from self-assembly of a stimuli-responsive amphiphilic block copolymer†

Abstract
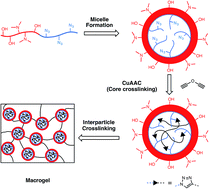
RAFT polymerization was utilized to prepare an amphiphilic block copolymer containing both hydrophilic and hydrophobic segments. The self-assembly behavior of the block copolymer into nano-scale particulate structures was studied in both water and polar organic solvents. Uniform micelle assemblies were stabilized by reaction within the hydrophobic core, which contained pendant azide groups, through copper-catalyzed azide-alkyne cycloaddition (CuAAC) click chemistry with a dialkyne crosslinker. The reaction preceded efficiently with negligible residual azide functionality and resulted in core–shell nanogel structures that were analyzed by a variety of techniques including light scattering, electron microscopy and the ability to take up hydrophobic molecules. Both thermo- and pH-responsive character of the nanogels and the linear polymers from which they were made were studied through cloud point testing at different pH levels. It was found that these nanogel dispersions in water exhibited the highest cloud point temperatures indicating a highly stable nanogel structure. The solvent-dispersed nanogels were used as building blocks to form extended polymer networks through the inter- as well as intra-particle reaction between hydroxyl groups within the hydrophilic domain of the nanogel shell by crosslinking with a diisocyanate. It was found that as little as 10 wt% nanogel dispersions in solvent reached the percolation threshold to yield highly porous macroscopic networks; while 50 wt% concentrations achieved densely packed and interdigitated nanogels to afford relatively homogeneous structures.
 Please wait while we load your content...
Please wait while we load your content...

