
Catalytic oxidation of NO by O2 over CeO2–MnOx: SO2 poisoning mechanism
Abstract
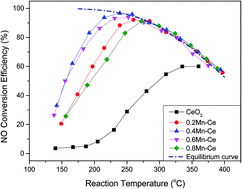
The catalytic oxidation of NO by O2 was performed over a series of CeO2–MnOx catalysts with different molar ratios of Mn/Ce, which were prepared by the sol–gel method. The highest NO conversion efficiency reached 96% over the catalyst with a 0.4 Mn doping value at 238 °C. The possible reaction pathways of the catalytic oxidation process were proposed according to several characterization measurements. NO was adsorbed on the catalyst surface to form nitrates and then decomposed into NO2. However, the catalyst was completely deactivated under an atmosphere of SO2. NO conversion efficiency dramatically declined from 92% to 22% within only 400 min. Comparing the BET, TPR, TPD, XRD, XPS, FTIR, and TGA results of fresh and poisoned catalysts, the catalyst deactivation could be mainly attributed to manganese sulfate formation on the catalyst surface, which could only slightly decompose. The active sites for NO adsorption were occupied. Finally, the oxidation of NO to NO2 was terminated due to lack of nitrates, which are intermediates for NO oxidation.
 Please wait while we load your content...
Please wait while we load your content...

