
From rice husk to high performance shape stabilized phase change materials for thermal energy storage
Abstract
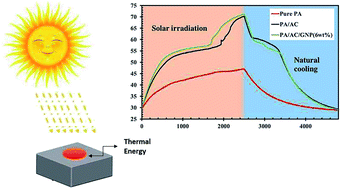
A novel shape-stabilized phase change material (SSPCM) was fabricated by using a vacuum impregnation technique. The lightweight, ultra-high specific surface area and porous activated carbon was prepared from waste material (rice husk) through the combination of an activation temperature approach and a sodium hydroxide activation procedure. Palmitic acid as a phase change material was impregnated into the porous carbon by a vacuum impregnation technique. Graphene nanoplatelets (GNPs) were employed as an additive for thermal conductivity enhancement of the SSPCMs. The attained composites exhibited exceptional phase change behavior, having a desirable latent heat storage capacity of 175 kJ kg−1. When exposed to high solar radiation intensities, the composites can absorb and store the thermal energy. An FTIR analysis of the SSPCMs indicated that there was no chemical interaction between the palmitic acid and the activated carbon with GNPs. The thermal conductivity of the prepared composites improved by more than 97% for the highest loading of GNPs (6 wt%) compared with that of pure palmitic acid. Moreover, the SSPCMs exhibit high thermal stability, with a stable melting–freezing enthalpy and excellent reversibility. The prepared SSPCMs with enhanced heat transfer and phase change properties provide a beneficial option for building energy conservation and solar energy applications owing to the low cost of raw materials and the simple synthetic technique.
 Please wait while we load your content...
Please wait while we load your content...

