
Ferrocene catalysed heteroarylation of BODIPy and reaction mechanism studies by EPR and DFT methods†
Swati Dixit,
Mahendra Patil* and
Neeraj Agarwal*
UM-DAE, Centre for Excellence in Basic Sciences, Health Center Building, Kalina Campus, Santacruz (E), Mumbai, 400098, India. E-mail: mahendra.patil@cbs.ac.in; na@cbs.ac.in
First published on 28th April 2016
Abstract
The C–H heteroarylations of BODIPy using heteroaryl diazoniumtetrafluoroborate and ferrocene (Fc) as catalysts were carried out. Mono and di heteroaryl BODIPy derivatives (1–8) were obtained in a cost-effective way. This method gives easy access to 4-pyridyl substituted BODIPy (7, 8), which is useful for synthesizing water soluble derivatives for biological applications. The pathway for this reaction was studied by carrying out spin cross-over experiments using electron paramagnetic resonance (EPR) techniques. A strong EPR signal was obtained in the presence of ferrocene and heteroaryldiazonium salt, which indicates the formation of radicals during the initial steps of the reaction. In addition, we have also performed computational investigations on different steps of the reaction to elucidate the role of ferrocene as a radical initiator in this reaction.
Introduction
4,4′-Difluoro-4-bora-3a,4a-diaza-s-indacene (BODIPy) is one of the most popular dye families and has been studied extensively in the past few years.1–3 BODIPy dyes are known for their rich optical properties and photostability. Photophysical, electrochemical and morphological properties of these dyes can be tuned, almost at will, which makes BODIPy an important fluorophore for applications in different areas.4–6 A variety of substituted BODIPys with variable degrees of conjugations have been synthesized to alter its photophysical properties, including multimeric BODIPy, conjugates of BODIPy with other fluorophores such as porphyrins and substitution with a variety of aryl groups.7 Heteroarylation of BODIPy has also been well explored, as substitution of heteroaryls changes the optical properties drastically.8 Even though many BODIPy derivatives bearing various heteroaryl substituents have been reported, methods of their synthesis are rather challenging. In general, to synthesize the heteroaryl derivatives of BODIPy one can start with the heteroaryl substituted pyrrole. A few reports are available for the synthesis of 3,5-diheteroaryl BODIPy in multiple steps.9 In some of these methods, 2-heteroaryl substituted pyrroles were first synthesized and further used in the synthesis of heteroaryl BODIPy. In other approaches, transition metal catalysed coupling reactions (Suzuki Miyaura and Stille coupling) were used to synthesize the aryl BODIPy derivatives.10 These coupling reactions require expensive transition metal catalyst, heteroaryl coupling partners and halogenated BODIPy. In these methods, purification of desired heteroaryl BODIPy is quite tedious.Transition-metal-free processes have been realized as alternative choices to perform various aryl C–C cross-coupling reactions. Several such reactions have been suggested, such as base promoted coupling reaction between unactivated arenes and aryl halides, which have been used in the presence of nitrogen heterocyclic ligands.11 In situ, metal complex formation with the ligand facilitates single-electron-transfer (SET) from metal tert-butoxides to ligands upon heating or in the presence of light.12 Light absorbing metal complexes (Ir-ppy complex) or other chromophores have been used as an active photoredox catalyst in C–H arylation.
Recently, Dehaen et al. showed a very useful, convenient, efficient and cost-effective method for C–H arylation of BODIPy13 in which phenyl BODIPy derivatives were synthesized by the reaction of phenyldiazoniumtetrafluoroborate with BODIPy in the presence of ferrocene as a catalyst. Later, alkylated BODIPy derivatives were synthesized using alkyltrifluoroborate or boronic acids and manganese acetate as a catalyst.14 Herein, we further extend the method developed by Dehaen et al. to synthesize heteroaryl substituted BODIPy derivatives (1–8) using a convenient and cost-effective way. Heteroaryl diazoniumtetrafluoroborate and ferrocene along with BODIPy were reacted in acetone at room temperature. To get insight on the possible reaction mechanism, electron paramagnetic resonance (EPR) experiments were carried out. Spin cross-over experiments with 5,5-dimethyl-pyrroline N-oxide (DMPO) in presence of ferrocene helped in trapping the radicals formed in the reaction. Furthermore, the reaction mechanism was investigated using density functional theory (DFT) methods. The comparison of free energies of different steps of the reaction, using ferrocene and other catalysts, such as CuCl and ascorbic acid, helped us to understand the role of ferrocene in this reaction. At this stage, we believe that reaction proceeds with radical formation. Absorption and emission properties of heteroaryl BODIPys are also reported.
Results and discussion
The heteroaryl BODIPys (1–8) were synthesized by the reaction of heteroaryl diazoniumtetrafluoroborate and BODIPy precursor with ferrocene as a catalyst (Scheme 1). The precursor 8-anisyl BODIPy was synthesized by the literature method,15 whereas the diazonium salts of heteroaryls were synthesized by a slightly modified classical method. The unsubstituted BODIPy was reacted with 2-thiadizole diazoniumtetrafluoroborate, 2-thiazole diazoniumtetrafluoroborate, 2-benzothiazole diazoniumtetrafluoroborate, and 4-pyridyl diazoniumtetrafluoroborate in the presence of ferrocene to obtain the compounds 1–8. All these reactions produced a mixture of mono- and di-heteroaryl substituted BODIPys. The exclusive synthesis of di-heteroaryl BODIPy could not be achieved, irrespective of the number of equivalents of diazonium salts. All these reactions were monitored by thin layer chromatography and absorption spectroscopy. After about 4 h, all the starting BODIPy was found to be consumed. Purification by column chromatography afforded the pure 1–8 in 40–70% yields except for compound 2. For compound 2 only a 4–5% yield was observed even in excess 2-thiadizole diazonium salt. Compounds 1–8 were characterized by several spectroscopic techniques, which include 1H-NMR, 13C-NMR, and MALDI-mass. Compounds 1–8 are fairly soluble in common organic solvents. | ||
| Scheme 1 Ferrocene catalysed synthesis of BODIPy derivatives 1–8. | ||
Fig. 1 shows the 1H-NMR and 13C-NMR of compound 6. Two doublets were obtained for pyrrolic protons between 8.0 and 8.2 ppm in 1H-NMR. These two doublets show the substitutions at the 3,5 positions only and not at the 2,6 positions, in which case we expect two singlets for pyrrolic protons. Substitutions at 3,5 positions was further confirmed with the help of correlation spectroscopy (COSY) (see ESI information†). Other peaks in the aromatic region were assigned to aryl substituents.
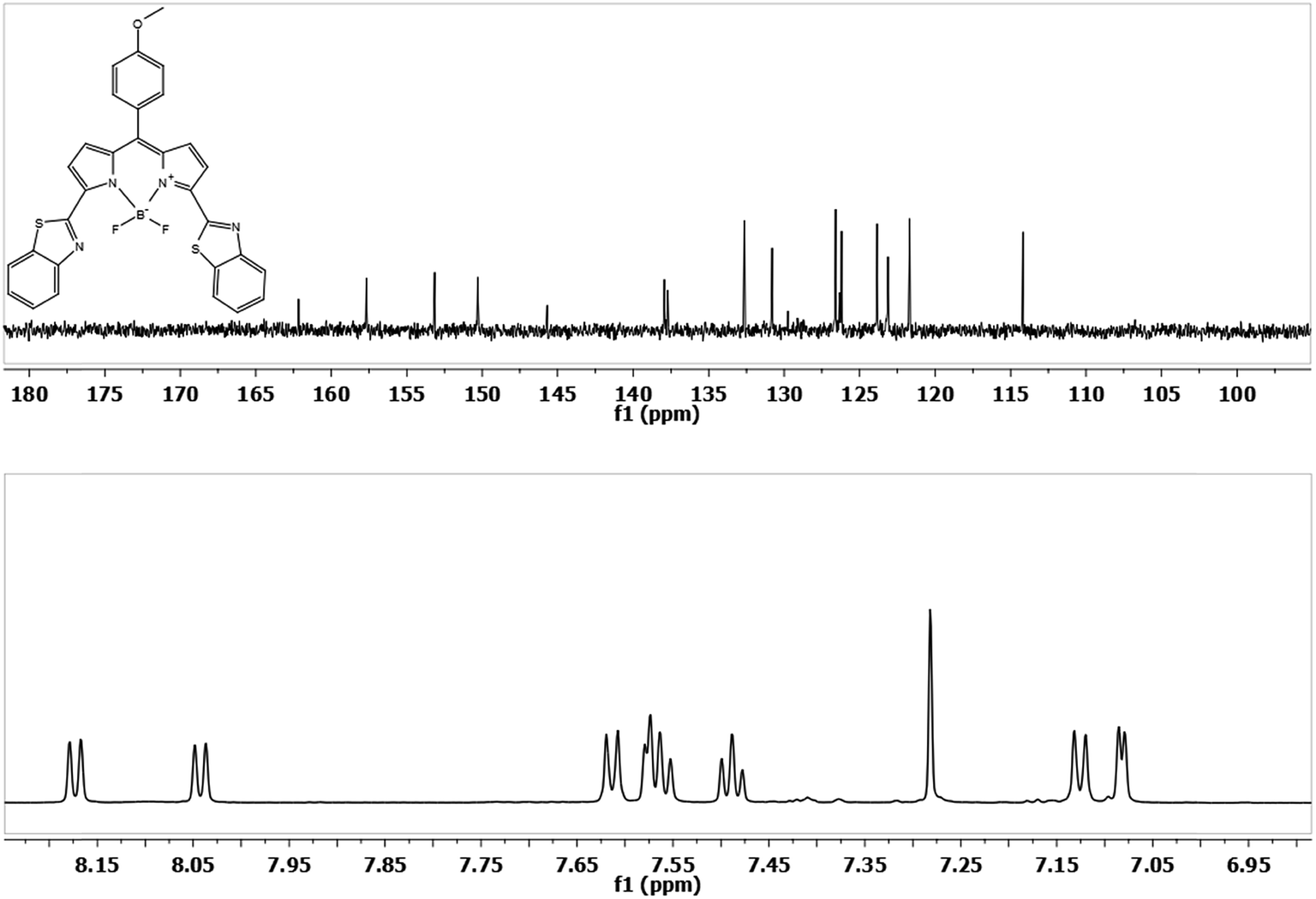 | ||
| Fig. 1 13C-NMR and 1H-NMR of BODIPy derivative 6 (only the aromatic region is shown for better clarity). | ||
To have an insight on the reaction mechanism, we carried out a set of electron paramagnetic resonance (EPR) experiments. The EPR spectra were obtained on an X band spectrometer (Bruker EMX series) at room temperature. Samples were degassed by passing high purity N2 gas before recording EPR. No signal was observed when the diazonium salt and ferrocene mixture was used. This does not rule out the absence of aryl radicals, as they may be short lived or efficiently relaxing. It is well known to use 5,5-dimethyl-pyrroline N-oxide (DMPO) as a spin trapping agent for aryl radicals in spin cross-over experiments.16 The reaction mixture of diazonium salt and DMPO produced a weak signal in EPR, which shows the presence of radical formation. A sharp increase in the intensity of this signal was observed when EPR was conducted with diazonium salt, DMPO and Fc together (Fig. 2). A sharp increase in the intensity of the EPR spectrum in the presence of Fc indicates the catalytic behaviour of Fc in the radical formation. The weak signal obtained in the absence of ferrocene was attributed to the self decomposition of diazonium salt. It is worth noting that the reaction of diazonium salt and BODIPy obtains the desired product even in the absence of ferrocene, albeit in lesser yield and with more reaction time.
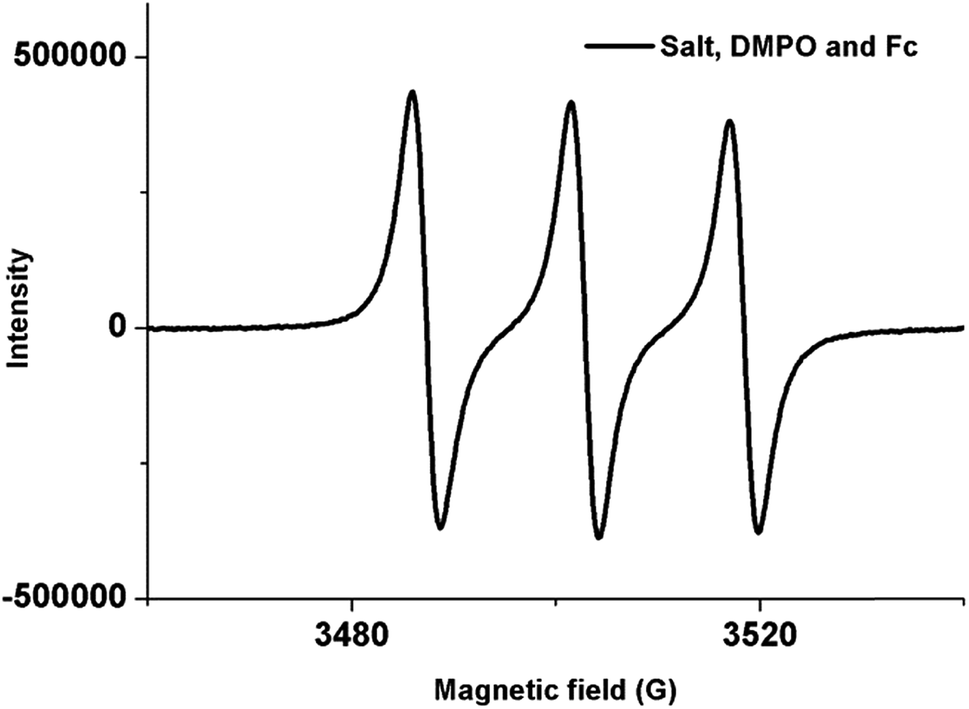 | ||
| Fig. 2 Electron paramagnetic resonance spectrum of 2-benzothiazolediazonium salt, ferrocene and DMPO in acetone. | ||
Having established the formation of radical intermediate in these reactions, we further investigated the reaction mechanism using density functional theory (DFT) calculations to establish the role of Fc in the reaction. The proposed steps involved in the reaction mechanism are shown in Scheme 2. All calculations were performed using Gaussian 09 suite of programs.17 The geometries of the reactants, intermediate structures and transition states were optimized in the solvent phase using the M06-2X functional18 (UM06-2X functional for open shell systems) with 6-311G** basis set. The solvent effect of acetone was taken into account by the polarizable continuum model (PCM).19 The free energy changes of each step of the reaction were calculated and provided in Table 1.
 | ||
| Scheme 2 Proposed reaction steps involved in the radical mechanism. | ||
| Step | Fc | CuCl | L-Ascorbic acid |
|---|---|---|---|
| Step-I (ΔG1) | −10.3 | 13.3 | 13.8 |
| Step-II (ΔG2) | −35.1 | — | — |
| Step-III (ΔG3) | −7.1 | −30.4 | −31.2 |
| Step-IV (ΔG4) | −12.1 | — | — |
The reaction commences with a single electron transfer (SET) from ferrocene to the aryl diazonium salt, which splits aryl diazonium salt into aryl radical and nitrogen molecule. The 2-thiazole diazoniumtetrafluoroborate was taken as a model aryl diazonium salt in the calculations. A SET from ferrocene to thiazole diazonium salt is found to be exoergic by 10.3 kcal mol−1. In the next step, thiazole radical adds to BODIPy to form the radical σ complex Ar-BODIPy˙. This step is predicted to be exoergic by 35.1 kcal mol−1. The transition state for the addition thiazole radical to BODIPy was also located at the M062X/6-311G** level of theory. The addition of thiozole radical to the BODIPy requires an activation barrier of 9.5 kcal mol−1. The resulting Ar-BODIPy˙ radical transfers an electron to the Fc+˙ to generate the cationic σ complex Ar-BODIPy+; this step is also found to be exoergic by 7.1 kcal mol−1. In final step, the Ar-BODIPy+ loses its proton to BF4− to provide heteroaryl BODIPy.
It is quite evident from these results that the ferrocene plays a crucial role in the electron transfer steps (step-I and step-III) because these steps are found to be fairly exoergic. To gain better insight on the role of ferrocene in electron transfer steps, free energies of these steps were calculated by replacing ferrocene with other reducing agents such as CuCl and ascorbic acid. The free energy changes of step-I and step-III involving CuCl and ascorbic acid are provided in Table 1. Interestingly, electron transfer from CuCl and ascorbic acid to thiazole diazonium salt is found to be endoergic by 13 kcal mol−1. Thus, it is clear that ferrocene ensures facile electron transfer to the diazonium salt and helps to initiate the aryl radical from diazonium salt.
BODIPy derivatives 1–8 were studied by absorption and emission spectroscopies in solution. Fig. 3 shows the absorption and emission spectra in toluene. Compounds 1–8 showed strong S0–S1 (π–π*) absorption maxima in the range of 524–632 nm. This band is assigned to the diisoindolomethene core of compounds 1–8. The absorption maximum of this band is directly dependent on the extent of de-localization of π electrons of heteroaryl substituents on BODIPy core. The broad band observed in the high energy region 414–461 nm of spectrum corresponds to the S0–S2 transition of the boradiazaindacene. Substitution of heteroaryl groups produced a large bathochromic shift of around 20–130 nm in absorption maxima in comparison to un-substituted BODIPy.15 Molar absorption coefficients for 1–8 were high and are summarized in Table 2.
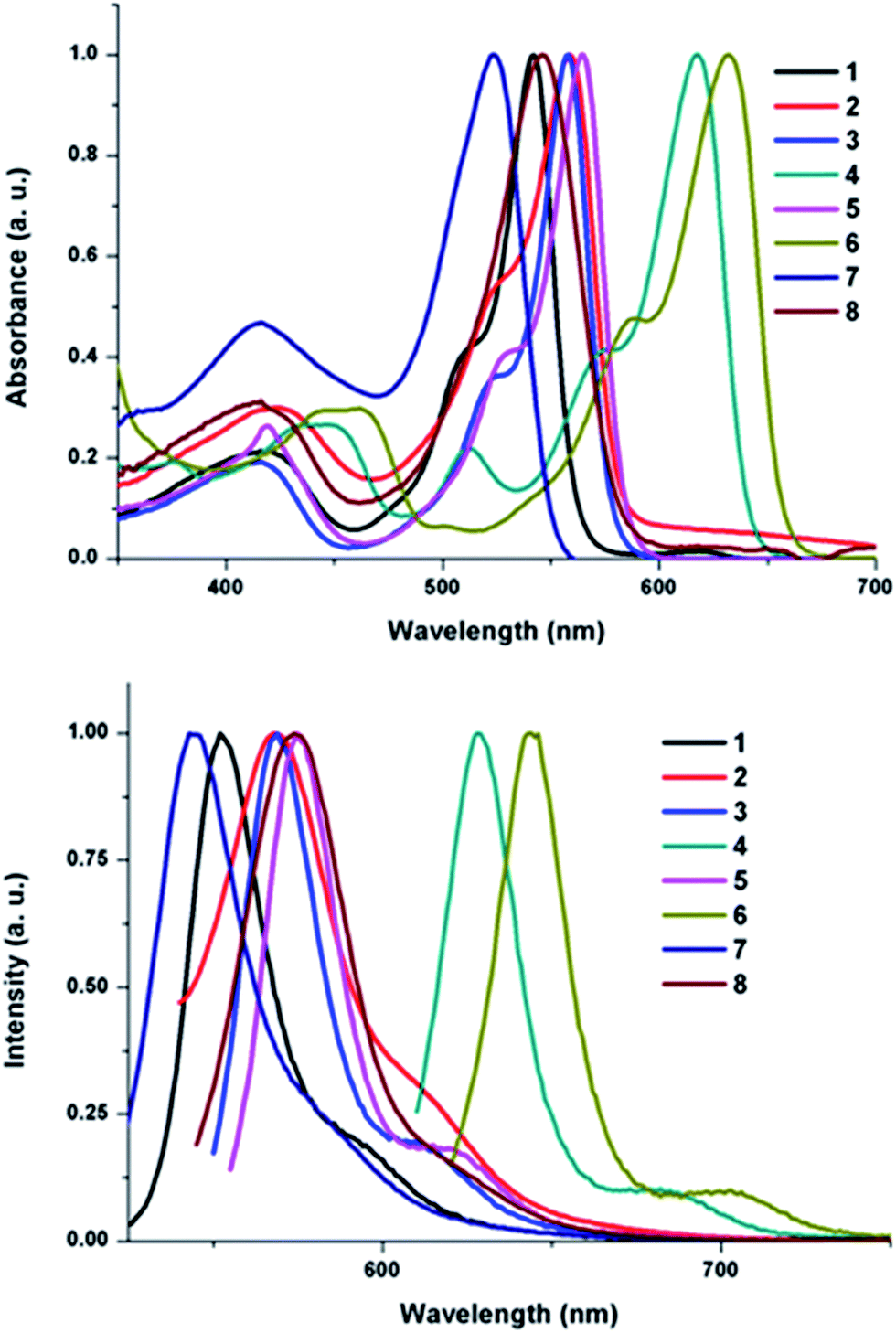 | ||
| Fig. 3 Absorption (above) and emission (below) spectra of 1–8 in toluene. | ||
| Compounds | λabs & ε [nm] (log) | λem [nm] | Φ | τ [ns] |
|---|---|---|---|---|
| a w.r.t. Rhodamine B.b w.r.t. tetraphenyl porphyrin. | ||||
| Std15 | 503 (4.73) | 521 | ||
| 1 | 542 (4.76), 509 (4.37), 414 (4.08) | 552, 594 | 65a | — |
| 2 | 559 (4.52), 523, 421 | 568 | 60a | — |
| 3 | 558 (4.87), 522 (4.42), 416 (4.15) | 568, 610 | 55a | 7.7 |
| 4 | 617 (4.83), 572 (4.45), 448 (4.26) | 628, 681 | 370b | 1.8 |
| 5 | 565 (4.95), 527 (4.54), 419 (4.33) | 575, 618 | 71a | 9.5 |
| 6 | 632 (4.85), 586 (4.52), 461 (4.32) | 644, 704 | 410b | 9.1 |
| 7 | 524 (4.67), 414 (4.46) | 544 | 22a | — |
| 8 | 546 (4.22), 414 (3.6) | 574 | 10a | — |
Emission spectra of compounds 1–8 recorded in toluene are shown in Fig. 3. Compounds 1–8 showed an emission peak at 544–644 nm with a low intensity shoulder in the further low energy region. Compounds 1–8 showed bathochromic shifts in emission spectra and up to 110 nm red shifts were observed in comparison to un-substituted BODIPy. A very small Stoke shift of ∼10 nm for 1–6 was observed, whereas for 7, 8 Stoke shift of ∼25 nm was observed, which suggests the change in the ground and excited state geometry is minimal in 1–6. Fluorescence quantum yields of compound 1–8 were calculated with respect to either Rhodamine B (ϕ = 0.68 in ethanol) or tetraphenyl porphyrin (TPP)20 (Table 2). Fluorescence life time studies of 3–6 were carried out in toluene and data are presented in Table 2.
Conclusion
The heteroarylation of BODIPy is achieved in moderate to good yields using diazonium tetrafluoroborate and ferrocene as a catalyst. Furthermore, the reaction mechanism was investigated using electron paramagnetic resonance (EPR) experiments and DFT calculations. Spin cross over experiments on the ferrocene mediated reaction showed sharp signals in EPR spectrum, indicating the formation of radical intermediate in the initial step of the reaction. We also elucidated the role of ferrocene as a radical initiator in this reaction using the DFT calculations. EPR experiments and DFT calculations support the radical mechanism for the ferrocene mediated heteroarylation of BODIPy using diazonium salt. The absorption and emission properties upon heteroaryl substitutions on BODIPy were studied. To the best of our knowledge, a conclusive proof for this mechanism is reported herein for the first time. We strongly believe that knowledge of the reaction mechanism pathway of this C–H arylation will help in designing advanced materials for various applications.Experimental section
Chemicals and instrumentation
All general chemicals and solvents were procured from S D. Fine Chemicals, India and Sigma-Aldrich. Column chromatography was performed using silica gel of 100–200 mesh size. The 1H and 13C-NMR (δ in parts per million) spectra were obtained using a Bruker 500 MHz spectrometer. Tetramethylsilane (TMS) was used as an internal reference for obtaining 1H-NMR spectra (residual proton; δ = 7.26 ppm) in CDCl3. The MS spectra were obtained on a Bruker MALDI-TOF. UV-Vis spectra were obtained on a Shimadzu 1800. Fluorescence measurements were carried out using a Horiba Fluoromax 4. For emission, compounds 1–8 were excited at 380 nm. Photophysical studies were carried out in toluene (∼1 × 10−6 M). The EPR spectra were obtained on an X band spectrometer (Bruker EMX series) at room temperature. Samples were degassed by passing high purity N2 gas before recording EPR.Synthesis and characterization of BODIPys (1–8)
A round bottom flask was charged with BODIPy (1 eqv.), respective aryl diazonium tetrafluoroborate (4 eqv.) and acetone. Ferrocene (0.1 eqv.) solution in acetone was added dropwise and slowly in 15 min time at room temperature. Reaction mixture was further stirred for 4 h. A noticeable colour change of reaction mixture was observed. Reaction mixture was extracted with dichloromethane and organic layers were washed with brine and water. Solvents were removed and crude product obtained was purified by column chromatography using hexane and dichloromethane to afford the compounds 1–8 (∼40–70% except for 2) as purple or greenish solids.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8) produced 1 as brown solid. Yield: (15 mg, 40%); mp: 132 °C; 1H-NMR (500 MHz, CDCl3): δ 9.32 (s, 1H), 8.03 (s, 1H), 7.60 (d, 2H, J = 8.55 Hz), 7.50 (d, 1H, J = 4.3 Hz), 7.11 (d, 2H, J = 8.5 Hz), 7.08 (t, 2H, J = 6.9 Hz), 6.68 (d, 1H, J = 3.6 Hz), 3.90 (s, 3H, OCH3); 13C-NMR (500 MHz, CDCl3): δ 154.05, 153.34, 145.45, 144.64, 139.27, 132.57, 132.49, 130.70, 126.01, 121.08, 114.27, 114.21; MALDI-TOF mass for C18H13BF2N4OS calcd 382.09, found 363 (M − F)+.:8) produced 1 as brown solid. Yield: (2 mg, 4%); mp: 168 °C; FT-IR (KBr, cm−1): ν 3090.6, 2920.7, 2851.4, 1706.9, 1525.6, 1546.3, 1503.5, 1395.9, 1370.9, 1297.2, 1253.2, 1176.8, 1127.3, 1069.4, 1021.2, 979.2, 879.4, 795.4, 739.9, 708.4, 608.1; MALDI-TOF mass for C20H13BF2N6OS2 calcd 466.63, found 446.63 (M − F)+.:8) produced 3 as brown solid. Yield: (20 mg, 52%); mp: 184 °C; FT-IR (KBr, cm−1): ν 3113.5, 2956.2, 2917.4, 1602.3, 1525.0, 1505.0, 1297.0, 1251.1, 1170.2, 796.8, 734.5; 1H-NMR (700 MHz, CDCl3): δ 8.04 (s, 1H), 7.97 (s, 1H), 7.66 (d, 1H, J = 3.85 Hz), 7.57 (d, 2H, J = 11.7 Hz), 7.35 (d, 1H, J = 5.3 Hz), 7.09 (d, 2H, J = 11.9 Hz), 7.04 (d, 1H, J = 5.2 Hz), 6.99 (s, 1H), 6.61 (s, 1H), 3.94 (s, 3H OCH3); 13C-NMR (600 MHz, CDCl3): δ 162.04, 157.41, 149.75, 144.55, 143.80, 137.26, 132.35, 130.55, 126.32, 123.74, 121.83, 113.98, 29.58; MALDI-TOF mass for C19H14BF2N3OS calcd 381.2, found 381 (M)+, 361.7 (M − F)+.:8) produced 4 as brownish purple solid. Yield: (25 mg, 54%); mp: 204.2 °C; FT-IR (KBr, cm−1): ν 3076.5, 2922.4, 2850.1, 1538.3, 1454.4, 1250.0, 1066.0, 853.6, 791.3, 734.5; 1H-NMR (700 MHz, CDCl3): δ 8.02 (d, 2H, J = 2.3 Hz), 7.64 (d, 2H, 2.85 Hz), 7.55 (d, 2H, J = 8.4 Hz), 7.37 (d, 2H, J = 3.9 Hz), 7.07 (d, 2H, J = 3.9 Hz), 6.98 (d, 2H, J = 8.4 Hz), 3.94 (s, 3H); 13C-NMR (500 MHz, CDCl3): δ 161.93, 157.48, 149.60, 146.16, 143.80, 143.49, 137.18, 134.76, 134.76, 132.36, 131.26, 130.70, 126.29, 123.42, 121.06, 118.83, 113.99, 29.57; MALDI-TOF mass for C22H15BF2N4OS2 calcd 464.32, found 463.9 (M)+, 444.9 (M − F)+.:8) produced 5 as brown-purple solid. Yield: (29 mg, 67%); mp: 179 °C; FT-IR (KBr, cm−1): ν 3116.4, 2918.3, 2844.5, 1548.4, 1496.3, 1392.8, 1128.3, 910.4, 838.4, 781.3, 723.7; 1H-NMR (500 MHz, CDCl3): δ 8.15 (d, 1H, J = 7.5 Hz), 8.02 (s, 1H), 7.97 (d, 1H, J = 7.3 Hz), 7.55–7.57 (m, 3H), 7.44 (s, 2H), 7.02–7.07 (m, 4H), 6.63 (s, 1H), 3.92 (s, 3H,–OCH3); 13C-NMR (700 MHz, CDCl3): δ 162.12, 157.82, 153.10, 149.04, 146.80, 144.73, 137.54, 137.26, 135.13, 132.50, 131.58, 130.76, 126.47, 126.25, 125.98, 123.73, 121.78, 121.53, 119.47, 114.10, 29.67; MALDI-TOF mass for C23H16BF2N3OS calcd 431.11, found 431.1 (M)+ and 411.9 (M − F)+.:8) produced 6 as brown solid. Yield: (37 mg, 66%); mp: 254 °C; FT-IR (KBr, cm−1): ν 3058.9, 2917.8, 2850.4, 1702.8, 1596.7, 1571.0, 1532.8, 1496.3, 1254.4, 1118.4, 1080.3, 1022.8, 879.1, 832.1; 1H-NMR (700 MHz, CDCl3): δ 8.17 (d, 2H, J = 8.12 Hz), 8.05 (d, 2H, J = 7.98 Hz), 7.62 (d, 2H, J = 8.33 Hz), 7.57–7.58 (m, 4H), 7.49 (t, 2H, J = 7.56 Hz), 7.12 (d, 2H, J = 8.26 Hz), 7.08 (d, 1H, J = 4.27 Hz), 3.96 (s, 3H, OCH3); 13C-NMR (500 MHz, CDCl3): δ 162.16, 157.66, 153.15, 150.29, 145.73, 137.94, 137.71, 132.63, 130.80, 129.75, 126.59, 126.20, 123.84, 123.11, 121.69, 114.19, 29.6; MALDI-TOF mass for C30H19BF2N4OS2 calcd 564.11, found 564.11 (M+) and 545.10 (M − F)+.:1) was used as eluent to get pure 8 as brownish purple solid. Yield: (18 mg, 46%); mp: 211 °C; 1H-NMR (700 MHz, CDCl3): δ 8.70 (d, 4H, J = 5.7 Hz), 7.70 (d, 4H, J = 5.75 Hz), 7.60 (d, 2H, J = 8.45 Hz), 7.11 (d, 2H, J = 8.55 Hz), 7.00 (d, 2H, J = 4.25 Hz), 6.7 (d, 2H, J = 4.2 Hz), 3.9 (s, 3H, OCH3); 13C-NMR (500 MHz, CDCl3): δ 155.43, 149.70, 139.65, 132.46, 131.75, 123.22, 120.76, 114.05, 107.44, 107.36, 96.11, 29.61; MALDI-TOF mass for C26H19BF2N4O calcd 452.26, found 452.34 (M)+.
:8) produced 1 as brown solid. Yield: (15 mg, 40%); mp: 132 °C; 1H-NMR (500 MHz, CDCl3): δ 9.32 (s, 1H), 8.03 (s, 1H), 7.60 (d, 2H, J = 8.55 Hz), 7.50 (d, 1H, J = 4.3 Hz), 7.11 (d, 2H, J = 8.5 Hz), 7.08 (t, 2H, J = 6.9 Hz), 6.68 (d, 1H, J = 3.6 Hz), 3.90 (s, 3H, OCH3); 13C-NMR (500 MHz, CDCl3): δ 154.05, 153.34, 145.45, 144.64, 139.27, 132.57, 132.49, 130.70, 126.01, 121.08, 114.27, 114.21; MALDI-TOF mass for C18H13BF2N4OS calcd 382.09, found 363 (M − F)+.:8) produced 1 as brown solid. Yield: (2 mg, 4%); mp: 168 °C; FT-IR (KBr, cm−1): ν 3090.6, 2920.7, 2851.4, 1706.9, 1525.6, 1546.3, 1503.5, 1395.9, 1370.9, 1297.2, 1253.2, 1176.8, 1127.3, 1069.4, 1021.2, 979.2, 879.4, 795.4, 739.9, 708.4, 608.1; MALDI-TOF mass for C20H13BF2N6OS2 calcd 466.63, found 446.63 (M − F)+.:8) produced 3 as brown solid. Yield: (20 mg, 52%); mp: 184 °C; FT-IR (KBr, cm−1): ν 3113.5, 2956.2, 2917.4, 1602.3, 1525.0, 1505.0, 1297.0, 1251.1, 1170.2, 796.8, 734.5; 1H-NMR (700 MHz, CDCl3): δ 8.04 (s, 1H), 7.97 (s, 1H), 7.66 (d, 1H, J = 3.85 Hz), 7.57 (d, 2H, J = 11.7 Hz), 7.35 (d, 1H, J = 5.3 Hz), 7.09 (d, 2H, J = 11.9 Hz), 7.04 (d, 1H, J = 5.2 Hz), 6.99 (s, 1H), 6.61 (s, 1H), 3.94 (s, 3H OCH3); 13C-NMR (600 MHz, CDCl3): δ 162.04, 157.41, 149.75, 144.55, 143.80, 137.26, 132.35, 130.55, 126.32, 123.74, 121.83, 113.98, 29.58; MALDI-TOF mass for C19H14BF2N3OS calcd 381.2, found 381 (M)+, 361.7 (M − F)+.:8) produced 4 as brownish purple solid. Yield: (25 mg, 54%); mp: 204.2 °C; FT-IR (KBr, cm−1): ν 3076.5, 2922.4, 2850.1, 1538.3, 1454.4, 1250.0, 1066.0, 853.6, 791.3, 734.5; 1H-NMR (700 MHz, CDCl3): δ 8.02 (d, 2H, J = 2.3 Hz), 7.64 (d, 2H, 2.85 Hz), 7.55 (d, 2H, J = 8.4 Hz), 7.37 (d, 2H, J = 3.9 Hz), 7.07 (d, 2H, J = 3.9 Hz), 6.98 (d, 2H, J = 8.4 Hz), 3.94 (s, 3H); 13C-NMR (500 MHz, CDCl3): δ 161.93, 157.48, 149.60, 146.16, 143.80, 143.49, 137.18, 134.76, 134.76, 132.36, 131.26, 130.70, 126.29, 123.42, 121.06, 118.83, 113.99, 29.57; MALDI-TOF mass for C22H15BF2N4OS2 calcd 464.32, found 463.9 (M)+, 444.9 (M − F)+.:8) produced 5 as brown-purple solid. Yield: (29 mg, 67%); mp: 179 °C; FT-IR (KBr, cm−1): ν 3116.4, 2918.3, 2844.5, 1548.4, 1496.3, 1392.8, 1128.3, 910.4, 838.4, 781.3, 723.7; 1H-NMR (500 MHz, CDCl3): δ 8.15 (d, 1H, J = 7.5 Hz), 8.02 (s, 1H), 7.97 (d, 1H, J = 7.3 Hz), 7.55–7.57 (m, 3H), 7.44 (s, 2H), 7.02–7.07 (m, 4H), 6.63 (s, 1H), 3.92 (s, 3H,–OCH3); 13C-NMR (700 MHz, CDCl3): δ 162.12, 157.82, 153.10, 149.04, 146.80, 144.73, 137.54, 137.26, 135.13, 132.50, 131.58, 130.76, 126.47, 126.25, 125.98, 123.73, 121.78, 121.53, 119.47, 114.10, 29.67; MALDI-TOF mass for C23H16BF2N3OS calcd 431.11, found 431.1 (M)+ and 411.9 (M − F)+.:8) produced 6 as brown solid. Yield: (37 mg, 66%); mp: 254 °C; FT-IR (KBr, cm−1): ν 3058.9, 2917.8, 2850.4, 1702.8, 1596.7, 1571.0, 1532.8, 1496.3, 1254.4, 1118.4, 1080.3, 1022.8, 879.1, 832.1; 1H-NMR (700 MHz, CDCl3): δ 8.17 (d, 2H, J = 8.12 Hz), 8.05 (d, 2H, J = 7.98 Hz), 7.62 (d, 2H, J = 8.33 Hz), 7.57–7.58 (m, 4H), 7.49 (t, 2H, J = 7.56 Hz), 7.12 (d, 2H, J = 8.26 Hz), 7.08 (d, 1H, J = 4.27 Hz), 3.96 (s, 3H, OCH3); 13C-NMR (500 MHz, CDCl3): δ 162.16, 157.66, 153.15, 150.29, 145.73, 137.94, 137.71, 132.63, 130.80, 129.75, 126.59, 126.20, 123.84, 123.11, 121.69, 114.19, 29.6; MALDI-TOF mass for C30H19BF2N4OS2 calcd 564.11, found 564.11 (M+) and 545.10 (M − F)+.:1) was used as eluent to get pure 8 as brownish purple solid. Yield: (18 mg, 46%); mp: 211 °C; 1H-NMR (700 MHz, CDCl3): δ 8.70 (d, 4H, J = 5.7 Hz), 7.70 (d, 4H, J = 5.75 Hz), 7.60 (d, 2H, J = 8.45 Hz), 7.11 (d, 2H, J = 8.55 Hz), 7.00 (d, 2H, J = 4.25 Hz), 6.7 (d, 2H, J = 4.2 Hz), 3.9 (s, 3H, OCH3); 13C-NMR (500 MHz, CDCl3): δ 155.43, 149.70, 139.65, 132.46, 131.75, 123.22, 120.76, 114.05, 107.44, 107.36, 96.11, 29.61; MALDI-TOF mass for C26H19BF2N4O calcd 452.26, found 452.34 (M)+.Acknowledgements
We thank Centre for Excellence in Basic Sciences, Mumbai for providing research facilities. Partial funding was provided by Department of Science and Technology, India (SR/FT/CS-87/2010). We thank Tata Institute of Fundamental Research, Mumbai for providing the NMR, EPR and mass facilities. We also thank Dr Yatendra Chaudhary, IMMT-Bhubaneshwar for providing the fluorescence decay facility.References
- A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891 CrossRef CAS PubMed.
- R. Ziessel, G. Ulrich and A. Harriman, Angew. Chem., Int. Ed., 2008, 47, 1184 CrossRef PubMed.
- V. Lakshmi, M. R. Rao and M. Ravikanth, Org. Biomol. Chem., 2015, 13, 2501 CAS.
- (a) A. B. Nepomnyashchii, M. Broering, J. Ahrens, R. Kruger and A. J. Bard, J. Phys. Chem. C, 2010, 114, 14453 CrossRef CAS; (b) C. Thivierge, A. Loudet and K. Burgess, Macromolecules, 2011, 44, 4012 CrossRef CAS PubMed.
- (a) S. Erten-Ela, M. D. Yilmaz, B. Icli, Y. Dede, S. Icli and E. U. Akkaya, Org. Lett., 2008, 10, 3299 CrossRef CAS PubMed; (b) J. C. T. Carlson, L. G. Meimetis, S. A. Hilderbrand and R. Weissleder, Angew. Chem., Int. Ed., 2013, 52, 6917 CrossRef CAS PubMed.
- (a) A. A. Rachford, R. Ziessel, T. Bura, P. Retailleau and F. N. Castellano, Inorg. Chem., 2010, 49, 3730 CrossRef CAS PubMed; (b) T. Tachikawa, N. Wang, S. Yamashita, S.-C. Cui and T. Majima, Angew. Chem., Int. Ed., 2010, 49, 8593 CrossRef CAS PubMed.
- (a) K. Rurack, M. Kollmannsberger and J. Daub, Angew. Chem., Int. Ed., 2001, 40, 385 CrossRef CAS; (b) Y. Yu, A. Descalzo, Z. Shen, H. Rohr, Q. Liu, Y. Wang, M. Spieles, Y. Li, K. Rurack and X. You, Chem.–Asian J., 2006, 1, 176 CrossRef CAS PubMed; (c) E. Fron, E. Coutino-Gonzalez, L. Pandey, M. Sliwa, M. Van der Auweraer, F. C. De Schryver, J. Thomas, Z. Dong, V. Leen, V. M. Smet, W. Dehaen and T. Vosch, New J. Chem., 2009, 33, 1490 RSC; (d) G. Ulrich, S. Goeb, A. De Nicola, P. Retailleau and R. Ziessel, J. Org. Chem., 2011, 76, 4489 CrossRef CAS PubMed; (e) T. Rohand, W. Qin, N. Boens and W. Dehaen, Eur. J. Org. Chem., 2006, 4658 CrossRef CAS; (f) M. Ravikanth, N. Agarwal and D. Kumaresan, Chem. Lett., 2000, 836 CrossRef CAS; (g) D. Kumaresan, N. Agarwal and M. Ravikanth, J. Chem. Soc., Perkin Trans. 1, 2001, 1644 RSC.
- (a) T. K. Khan, S. K. Jana, M. R. Rao, M. S. Shaikh and M. Ravikanth, Inorg. Chim. Acta, 2012, 383, 257 CrossRef CAS; (b) V. Leen, T. Leemans, N. Boens and W. Dehaen, Eur. J. Org. Chem., 2011, 4386 CrossRef CAS; (c) T. Rohand, W. Qin, N. Boens and W. Dehaen, Eur. J. Org. Chem., 2006, 4658 CrossRef CAS; (d) K. Kim, S. H. Choi, J. Jeon, H. Lee, J. O. Huh, J. Yoo, J. T. Kim, C.-H. Lee, Y. S. Lee and D. G. Churchill, Inorg. Chem., 2011, 50, 5351 CrossRef CAS PubMed.
- (a) Y. M. Poronik, V. P. Yakubovskyi, M. P. Shandura, Y. G. Vlasenko, A. N. Chernega and Y. P. Kovtun, Eur. J. Org. Chem., 2010, 2746 CrossRef CAS; (b) E. Yu. Schmidt, B. A. Trofimov, A. I. Mikhaleva, N. V. Zorina, N. I. Protzuk, K. B. Petrushenko, I. A. Ushakov, M. Yu. Dvorko, R. Méallet-Renault, G. Clavier, T. T. Vu, H. T. T. Tran and R. B. Pansu, Chem.–Eur. J., 2009, 15, 5823 CrossRef CAS PubMed.
- (a) V. Lakshmi and M. Ravikanth, J. Org. Chem., 2013, 78, 4993 CrossRef CAS PubMed; (b) M. Vincent, E. Beabout, R. Bennett and P. Hewavitharanage, Tetrahedron Lett., 2013, 54, 2050 CrossRef CAS; (c) I. Esnal, J. Banuelos, I. Lopez Arbeloa, A. Costela, I. García-Moreno, M. Garzon, A. R. Agarrabeitia and M. Jose Ortiz, RSC Adv., 2013, 3, 1547 RSC; (d) P. Gautam, B. Dhokale, S. M. Mobin and R. Misra, RSC Adv., 2012, 2, 12105 RSC; (e) A. Coskun and E. U. Akkaya, Tetrahedron Lett., 2004, 45, 4947 CrossRef CAS; (f) G. Ulrich, C. Goze, S. Goeb, P. Retailleau and R. Ziessel, New J. Chem., 2006, 30, 982 RSC; (g) V. P. Yakubovskyi, M. P. Shandura and Y. P. Kovtun, Eur. J. Org. Chem., 2009, 3237 CrossRef CAS; (h) J.-Y. Liu, H. S. Yeung, W. Xu, X. Li and D. K. P. Ng, Org. Lett., 2008, 10, 5421 CrossRef CAS PubMed; (i) D. Lakhe, K. K. Jairaj, M. Pradhan, U. Ladiwala and N. Agarwal, Tetrahedron Lett., 2014, 55, 7124 CrossRef CAS.
- (a) S. Yanagisawa, K. Ueda, T. Taniguchi and K. Itami, Org. Lett., 2008, 10, 4673–4676 CrossRef CAS PubMed; (b) W.-C. Chen, Y.-C. Hsu, W.-C. Shih, C.-Y. Lee, W.-H. Chuang, Y.-F. Tsai, P. P.-Y. Chen and T.-G. Ong, Chem. Commun., 2012, 48, 6702–6704 RSC; (c) Y. Cheng, X. Gu and P. Li, Org. Lett., 2013, 15, 2664–2667 CrossRef CAS PubMed; (d) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC.
- (a) M. E. Budén, J. F. Guastavino and R. A. Rossi, Org. Lett., 2013, 15, 1174–1177 CrossRef PubMed; (b) Y. Cheng, X. Gu and P. Li, Org. Lett., 2013, 15, 2664–2667 CrossRef CAS PubMed.
- B. Verbelen, S. Boodts, J. Hofkens, N. Boens and W. Dehaen, Angew. Chem., Int. Ed., 2015, 54, 4612 CrossRef CAS PubMed.
- B. Verbelen, L. C. D. Rezende, S. Boodts, J. Jacobs, L. V. Meervelt, J. Hofkens and W. Dehaen, Chem.–Eur. J., 2015, 21, 12667 CrossRef CAS PubMed.
- R. W. Wagner and J. S. Lindsey, Pure Appl. Chem., 1996, 68, 1373 CrossRef CAS.
- A. Bosnjakovic and S. Schlick, J. Phys. Chem. B, 2004, 108, 4332 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox, Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215 CrossRef CAS.
- (a) M. Cossi, V. Barone, R. Cammi and J. Tomasi, Chem. Phys. Lett., 1996, 255, 327–335 CrossRef CAS; (b) E. Cances, B. Mennucci and J. Tomasi, Chem. Phys., 1997, 107, 3032 CAS; (c) M. Cossi, G. Scalmani, N. Rega and V. Barone, Chem. Phys., 2002, 117, 43 CAS.
- R. F. Kubin and A. N. Fletcher, J. Lumin., 1983, 27, 455 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra03705d |
| This journal is © The Royal Society of Chemistry 2016 |