DOI:
10.1039/C6RA03694E
(Paper)
RSC Adv., 2016,
6, 40750-40759
Co-sensitization of “H”-type dyes with planar squaraine dyes for efficient dye-sensitized solar cells†
Received
9th February 2016
, Accepted 11th April 2016
First published on 13th April 2016
Abstract
Three “H”-type dyes with different linkage mode of two “D–π–A” moieties (LI-101, LI-102 and LI-103) were synthesized and applied to the co-sensitized system with the famous metal-free NIR squaraine dye of SQ2. Thanks to the advantage of the “H” configuration and their complementary spectra, the co-sensitized system exhibited the much improved photovoltaic performance of about a 70% enhancement, in comparison with that of SQ2 itself. Among them, the LI-102/SQ2 co-sensitized solar cell exhibited the highest conversion efficiency of 7.44% with 16.6 mA cm−2 for Jsc, 714 mV for Voc, 0.63 for FF.
Introduction
Nowadays, dye-sensitized solar cells (DSCs) have gained increasing attention from both industry and academic research, since they present many potential advantages such as flexibility, lightweight, low cost and easy processing with colorful and transparent features, which render them greatly promising for the next generation of photovoltaic devices.1–3 In this type of nanostructured photovoltaic cells, a self-assembled monolayer of dye molecules plays a key role by harvesting the light and injecting the photogenerated electron into the semiconductor oxide (TiO2). Apart from the zinc porphyrin and ruthenium polypyridine complexes,4,5 considerable efforts have also been devoted to the design and synthesis of numerous metal-free organic dyes,6–10 primarily due to the abundance of raw materials, flexibility of molecule design, and the low cost. Generally, the organic dyes exhibited relatively narrow absorption in the UV-visible region, directly limiting the development of the corresponding photovoltaic devices. Though many efforts have been made by tuning the structures of organic dyes, it is still a challenge to catch the rainbow by a single small molecule dye.11,12 Alternatively, multiple dyes with complementary spectral profiles used as “dye cocktails” for co-sensitization pave a new approach to realize a UV-visible-NIR spectral response with preferable photovoltaic performances.13–15
Notably, squaraine (SQ) based dyes have been widely applied as NIR photosensitizers because of their photolytic stability and extraordinary intense absorption in the NIR spectral region with high ε value at the level of 105 M−1 cm−1.16–20 However, the photovoltaic performances of SQ-based DSCs were not as good as expected, mainly due to the following two factors. One is the absent absorption below 500 nm, which leads to the poor light-harvesting ability, the other is the formation of aggregates on the TiO2 surface that induces the quenching of photo-excited states and quick charge recombination, resulting in the lower Voc values. According to the merits of SQ2 dye and co-sensitized systems, some groups had researched the co-sensitized system for SQ2 dye. Ho et al. used organic dyes of BP-1, BP-2 and BP-3 (Chart S1†) in combination with SQ2, to give a higher efficiency (8.14%) of the BP2/SQ2 co-sensitized system than the single ones (BP-2, 5.95%; SQ2, 3.78%).17 Liu et al. fabricated dye-sensitized solar cells based on indoline dye D131 (Chart S1†) and SQ2, with an efficiency of 4.1% on plastic substrates.18 Ho et al. raised the efficiency from 5.44% (a diarylaminofluorene-based organic dye, JD1 (Chart S1†)) to 6.36% (JD1 and SQ2).19 Recently, Cooke et al. reported co-sensitized DSCs with half-squarylium dye 7b (Chart S1†) and squaraine dye SQ2 (Chart 1) with an efficiency of 6.1% for co-sensitized cells, as compared to 7b dye (5.0%) and SQ2 dye (3.4%) themselves.20 With the aim to remedy the disadvantages of SQ2 and further explore new co-sensitized systems, the organic dyes with UV-visible absorption and stereoscopic structures should be employed as the co-sensitizers.
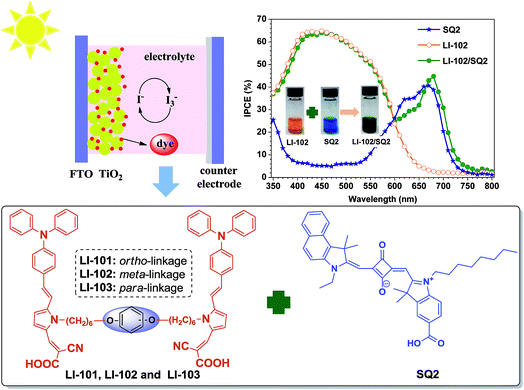 |
| | Chart 1 The co-sensitized system consisted of LI-101, LI-102, LI-103 and SQ2. | |
“H”-type sensitizers, first proposed by our group, were a class of organic dyes with two pieces of D–π–A (donor-π bridge-acceptor) chromophore moieties sharing one isolation group,21 which have been proved as a novel structure to increase the dye-loading amount and suppress the dye aggregates, leading to the improved device performance. Moreover, apart from the thiophene unit as the common conjugated bridge, the seldom used one, pyrrole ring, exhibited some unique properties, such as the electron-rich character and the multiple active sites, which were beneficial to the intramolecular charge transfer (ICT) and the synthesis of “H”-structures, resulting in the higher light-harvesting abilities. Therefore, considering the advantage of pyrrole-based sensitizers with “H”-type and planar squaraine dye with NIR absorption, in this paper, we synthesized three N-alkylpyrrole based “H” type dyes (LI-101, LI-102 and LI-103, Chart 1) with different linkage modes of the two D–π–A chromophore moieties, with the aim to tune the opto-electronic properties of “H”-type sensitizers subtly. Fortunately, once utilized for the co-sensitization with SQ2, the enhanced conversion efficiencies were obtained than that of the single ones. Among them, the solar cell co-sensitized by dye LI-102 with meta-linkage model and SQ2 showed the best photovoltaic performance, with Jsc of 16.6 mA cm−2, Voc of 714 mV, FF of 0.63 and η of 7.44%, much higher than that of SQ2 itself (4.28%). Herein, we would like to report their syntheses, structural characterization, electrochemical properties, theoretical calculations and photovoltaic performance.
Results and discussion
Synthesis
The synthetic route to LI-101, LI-102 and LI-103 was depicted in Scheme 1. Compound 2a, 2b and 2c were prepared by the nucleophilic substitution reaction to construct the different linkage mode between the pyrrole ring and the middle dialkoxyphenyl unit. Through the following Wittig reaction with diethyl 4-(diphenylamino)benzylphosphonate for the introduction of the electron donor, compound 3a, 3b and 3c were yielded respectively. Due to the high reactivity and electron-rich property of pyrrole ring, the aldehydes 4a, 4b and 4c were obtained by the normal Vilsmeier reaction with relatively high yields. Finally, the organic dyes of LI-101, LI-102 and LI-103 were prepared from the corresponding aldehydes and cyanoacetic acid through the Knoevenagel reaction, in the presence of piperidine as catalyst. The structure and purity of the target molecules were confirmed by the standard spectroscopic methods.
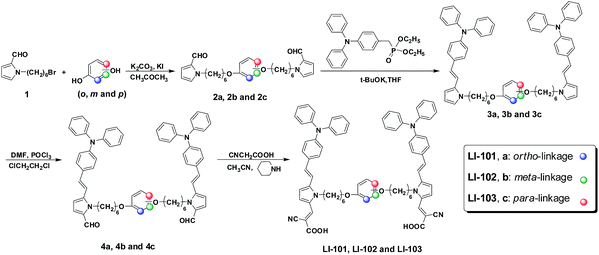 |
| | Scheme 1 The synthetic route of LI-101–LI-103. | |
Optical properties
Fig. 1 showed the absorption spectra of the dyes in a mixture solution of acetonitrile and t-BuOH (volume ration of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), with the corresponding photophysical data listed in Table 1. In comparison to the reference dye LI-54 with single D–π–A structure (Chart S2 in ESI†), dye LI-101, LI-102 and LI-103 exhibited a boarder absorption band in the region of 400–600 nm, along with the red-shift (about 50 nm) of the maximum absorption wavelength (λmax), which were mainly due to the interaction of the two D–π–A moieties in the “H” structure and the strong intramolecular charge transfer (ICT) from the electron donor part (D) to the electron acceptor (A). Moreover, the molar extinction coefficients (ε) of the “H”-type dyes were almost as high as two times that of the single one (LI-54), due to the two D–π–A moieties per H-type molecule. And this configuration could be beneficial to forming the compact layer on the TiO2 surface, resulting in the high light-harvesting ability.22 Regardless of the different linkage modes of the three “H”-type dyes, their absorption spectra were similar to each other, owing to the same D–π–A moieties. On the other hand, in the wavelength range of 550–700 nm, SQ2 exhibited an intense absorption, which was just complementary with that of dye LI-101, LI-102 and LI-103, well realizing the UV-visible-NIR spectral response (Fig. 1). In the solutions consisting of “H” type dyes and SQ2, the corresponding absorption spectra became almost sequential from 350 to 700 nm, and “H” type dyes still exhibited higher light-harvesting abilities than that of LI-54 in this mixture system. When adsorbed on a transparent thin TiO2 film, the absorption spectra became broader than those in solution, which should be attributed to the interactions between the dyes and TiO2 films, reported in the literatures.23 Interestingly, in the case of co-sensitized films, the contribution of the four dyes (LI-101, LI-102, LI-103 and LI-54) to the absorption in the region of 350–600 nm were different in a large degree, and the “H” type dyes still showed better performance, which varied with the changeable linkage modes. As to dye LI-102 with the meta-linkage mode, the absorption in the UV-visible region achieved the maximum, indicating that the different “H” structures led to the various alignments on the TiO2 surface, and the configuration of LI-102 may be favorable to the regular arrangement of the sensitizers on the TiO2 surface.
:1), with the corresponding photophysical data listed in Table 1. In comparison to the reference dye LI-54 with single D–π–A structure (Chart S2 in ESI†), dye LI-101, LI-102 and LI-103 exhibited a boarder absorption band in the region of 400–600 nm, along with the red-shift (about 50 nm) of the maximum absorption wavelength (λmax), which were mainly due to the interaction of the two D–π–A moieties in the “H” structure and the strong intramolecular charge transfer (ICT) from the electron donor part (D) to the electron acceptor (A). Moreover, the molar extinction coefficients (ε) of the “H”-type dyes were almost as high as two times that of the single one (LI-54), due to the two D–π–A moieties per H-type molecule. And this configuration could be beneficial to forming the compact layer on the TiO2 surface, resulting in the high light-harvesting ability.22 Regardless of the different linkage modes of the three “H”-type dyes, their absorption spectra were similar to each other, owing to the same D–π–A moieties. On the other hand, in the wavelength range of 550–700 nm, SQ2 exhibited an intense absorption, which was just complementary with that of dye LI-101, LI-102 and LI-103, well realizing the UV-visible-NIR spectral response (Fig. 1). In the solutions consisting of “H” type dyes and SQ2, the corresponding absorption spectra became almost sequential from 350 to 700 nm, and “H” type dyes still exhibited higher light-harvesting abilities than that of LI-54 in this mixture system. When adsorbed on a transparent thin TiO2 film, the absorption spectra became broader than those in solution, which should be attributed to the interactions between the dyes and TiO2 films, reported in the literatures.23 Interestingly, in the case of co-sensitized films, the contribution of the four dyes (LI-101, LI-102, LI-103 and LI-54) to the absorption in the region of 350–600 nm were different in a large degree, and the “H” type dyes still showed better performance, which varied with the changeable linkage modes. As to dye LI-102 with the meta-linkage mode, the absorption in the UV-visible region achieved the maximum, indicating that the different “H” structures led to the various alignments on the TiO2 surface, and the configuration of LI-102 may be favorable to the regular arrangement of the sensitizers on the TiO2 surface.
 |
| | Fig. 1 Absorption spectra of dye LI-101, LI-102, LI-103 and LI-54 in ACN/t-BuOH (1:1) solution (A) and in TiO2 films (4 μm) (C); absorption spectra of co-sensitizer LI-101/SQ2, LI-102/SQ2, LI-103/SQ2, LI-54/SQ2 and SQ2 in ACN/t-BuOH (1:1) solution (B) and in TiO2 films (4 μm) (D). | |
Table 1 Opto-electronic properties of organic dyes
| Dye |
λmaxa (nm) |
ε (105 M−1 cm−1) |
E0–0b (eV) |
ED/D+c (V) |
ED*/D+d (V) |
LUMOe (eV) |
HOMOf (eV) |
| Absorption maximum of the dyes measured in acetonitrile/t-BuOH (1:1); ε: molar extinction coefficient at λmax. E0–0 was determined from the intersection of the tangent of absorption on the TiO2 film and the x axis by 1240/λonset. The ground-state redox potential (ED/D+) were measured in CH2Cl2 with 0.1 M (n-C4H9)4NPF6 as electrolyte (scanning rate, 100 mV s−1; working electrode and counter electrode, Pt wires; reference electrode, Ag/AgCl). ED*/D+ was calculated from ED/D+ − E0–0. The HOMO was taken from the first redox potential in CV plot. The LUMO was calculated with the expression of LUMO = HOMO − E0–0. |
| LI-101 |
508 |
0.95 |
2.19 |
0.95 |
−1.24 |
−2.88 |
−5.07 |
| LI-102 |
516 |
1.05 |
2.18 |
0.95 |
−1.23 |
−2.89 |
−5.07 |
| LI-103 |
510 |
0.98 |
2.22 |
0.96 |
−1.26 |
−2.86 |
−5.08 |
| LI-54 |
452 |
0.63 |
2.21 |
0.90 |
−1.31 |
−2.99 |
−5.00 |
Electrochemical properties
In order to judge the possibilities of the electron injection from the excited dye molecules to the conduction band of TiO2 and the dye regeneration of dye sensitizers, the energy levels were investigated by cyclic voltammograms (Fig. S1†), with the electrochemical data of the three “H” type dyes summarized in Table 1. The ground-state redox potentials (ED/D+) of the three dyes, corresponding to the HOMO levels, were similar and located at about 0.95 V. The results suggested that the different linkage positions of the two D–π–A moieties in the “H” type dyes did not influence electrochemical properties of the whole conjugated system, and the electron transfers in the D–π–A moieties were still similar to that of the single one (LI-54). The HOMO levels of these dyes were more positive than the redox potential of the I−/I3− redox couple (∼0.4 V vs. NHE), ensuring the driving force for the regeneration of oxidized dye molecules efficiently. On the basis of the zero–zero transition energies (E0–0) estimated from the observed optical edge, we could calculate the excited state redox potential via the equation ED*/D+ = ED/D+ + E0–0/e, being −1.24, −1.23 and −1.26 V for LI-101–LI-103, respectively, which did not have much difference either. The ED*/D+ of dyes corresponded to the lowest unoccupied molecular orbital (LUMO) levels, which were more negative than the conduction band of TiO2 electrode (−0.5 V vs. NHE), ensuring sufficient driving force of these three dyes for the electron injection from the excited sensitizers to TiO2 surface (Fig. 2). Thus, the three “H” dyes were energetically permitted to function in DSCs.
 |
| | Fig. 2 Energy-level diagram of sensitizers, electrolyte and TiO2. ΔG1: driving force for electron injection; ΔG2: driving force for regeneration of the oxidized dyes. | |
Theoretical approach
To gain further insight into the correlation between molecular structure and electron distribution, the dye molecules were optimized by density functional theory (DFT) calculations at the B3LYP/6-31G* level with Gaussian 09 program.24 The optimized structures and the electronic distributions in HOMO and LUMO were shown in Fig. 3. As easily seen, in the three “H” type dyes, the two pieces of D–π–A moieties had planar geometries, and the dihedral angles of the triphenylamine–vinylene–pyrrole were much smaller, indicating that the middle linkage units as the isolation group did not twist the D–π–A conjugated system. Thus, this “H” configuration could not only keep the charge transfer through the whole conjugation system, but also prevent the π–π* stacking between the planar structures. However, with the different linkage modes, the topological structures of the three dyes varied largely. The ortho-linkage mode made the structure of dye LI-101 twist in a large degree, and the two pieces of the D–π–A moieties were almost perpendicular. And the configurations of LI-102 and LI-103 were more similar to the shape of “H”. These different topological structure would surely affect their absorption and alignment on the TiO2 surface, as supported by the absorption spectra of the three dyes (Fig. 1). The electronic distributions in HOMO and LUMO of the three dyes were similar. The electronic distributions in HOMO mainly located on the TPA donor and pyrrole unit (the conjugated bridge) and the LUMOs were distributed on the conjugated bridges and cyanoacetic acid system. The well over-lapped HOMO and LUMO orbits on the pyrrole units allowed an efficient intramolecular charge transfer, which were favorable for the light-harvesting and electron transitions.
 |
| | Fig. 3 Frontier orbitals of the dyes optimized at the B3LYP/6-31G* level. | |
Photovoltaic performance of DSCs
DSCs with an effective area of 0.25 cm2 were fabricated by a sandwich structure comprising of 0.6 M DMPII (1,2-dimethyl-3-propylimidazolium iodide), 0.1 M LiI, 0.03 M I2, 0.5 M 4-TBP (4-tert-butylpyridine) in acetonitrile/3-methoxypropionitrile (MPN) (1:1, v/v) as the redox electrolyte. In the Experimental section, the details of the device preparation and characterization were described. The incident photon-to-current conversion efficiencies (IPCE) of the cells based on single dyes and co-sensitization were investigated, which could be used to estimate the light harvesting efficiency and short circuit photocurrent generation capability of the DSCs (Fig. 4). All the three “H” type sensitizers (LI-101, LI-102 and LI-103) exhibited broader IPCE curves than that of the normal one (LI-54), with the conversion of the light to photocurrent in the region from 350 to 650 nm. After the co-sensitization with NIR dye SQ2, the IPCE curves can extend to 750 nm to harvest more sunlight, due to the complementary spectra of the “H” type dyes and SQ2. As shown in Fig. 4, the much lower IPCE of SQ2 in the region of 400–550 nm (below 10%) could be increased to above 60% by incorporating one of the three organic dyes with “H” structure, agreeing well with their absorption spectra in the TiO2 film.
 |
| | Fig. 4 Spectra of monochromatic IPCE for DSCs based on the single dyes and co-sensitized system. | |
The J–V plots of DSCs based on the single sensitizer and co-sensitized system were shown in Fig. 5 and S2,† and the corresponding parameters were presented in Tables 2 and S1.† As to the organic dyes with one D–π–A unit (LI-54) and “H” structures (LI-101, LI-102 and LI-103), the better photovoltaic performance of the “H” type dyes sensitized solar cells was obvious, mainly due to the higher Jsc values. The results indicated that “H” type dyes bearing two D–π–A moieties in a suitable distance would be favorable to the light harvesting, as the key factors to Jsc. By changing the linkage modes of the two D–π–A pieces on a benzene ring from LI-101 to LI-103, the photovoltaic performance of three H-type dyes varied in some degree. For the most twisted structure of LI-101, the lowest Voc (692 mV) was generated for the possible charge recombination on the dye/TiO2/electrolyte interface. Since the similar dye loading amounts of the three “H” dyes (Table 2), the highest Jsc value of LI-102 (12.95 mA cm−2) may be due to the relatively regular “H” structure, which was beneficial to the alignment of dyes on the TiO2 surface and the more compact dye layer.
 |
| | Fig. 5 J–V characteristics of DSCs measured at simulated 100 mW cm−2 AM 1.5 conditions. | |
Table 2 Performance data of DSCs based on single dyes and co-sensitized system
| Sensitizer |
Jsc (mA cm−2) |
Voc (mV) |
FF |
η (%) |
Rrec (Ω cm−2) |
τ (ms) |
Dye loading amount (10−7 mol cm−2) |
| The TiO2 electrode was stained by immersing it into the single dye solution (0.3 mM) in acetonitrile/tert-butanol (1:1, v/v) for 24 h. The TiO2 electrode was stained by immersing it into the mixture of the two dyes (0.3 mM) and CDCA (5 mM) in acetonitrile/tert-butanol (1:1, v/v) for 24 h. |
| LI-101a |
12.70 |
692 |
0.63 |
5.58 |
57.95 |
22.51 |
2.72 |
| LI-102a |
12.95 |
717 |
0.64 |
5.98 |
112.70 |
35.54 |
2.20 |
| LI-103a |
12.50 |
715 |
0.64 |
5.75 |
78.26 |
23.14 |
2.60 |
| LI-54a |
10.79 |
690 |
0.72 |
5.39 |
55.13 |
22.09 |
3.15 |
| SQ2b |
9.88 |
632 |
0.69 |
4.28 |
24.68 |
11.16 |
1.01 |
| LI-101/SQ2b |
16.47 |
685 |
0.63 |
7.10 |
87.23 |
22.71 |
1.01, 0.53 |
| LI-102/SQ2b |
16.62 |
714 |
0.63 |
7.44 |
95.38 |
35.15 |
1.80, 0.49 |
| LI-103/SQ2b |
15.98 |
718 |
0.63 |
7.19 |
117.80 |
38.93 |
1.67, 0.29 |
| LI-54/SQ2b |
12.68 |
620 |
0.71 |
5.62 |
34.38 |
9.80 |
2.10, 0.46 |
As to the NIR dye SQ2 with planar structure, the low conversion efficiency (η) of 2.76% was obtained with a Jsc of 6.66 mA cm−2, a Voc of 597 mV, and a FF of 0.69 (Fig. S2 and Table S1†). After the common nonchromophoric co-adsorbent,25–28 CDCA (chenodeoxycholic acid), was incorporated to suppress the possible interactions between the adjacent dyes, an improved photovoltaic performance was achieved with an increased η of 4.28%, partially hinting that the severe aggregates of SQ2 dyes on the TiO2 surface. For the organic dyes with “H” structure, the addition of CDCA in the fabrication process, affected little on the photovoltaic performance (Fig. S2 and Table S1†), suggesting their anti-aggregated properties in a large degree. Then, the co-sensitization of the “H” type dyes (LI-101, LI-102 and LI-103) and SQ2 would be not only helpful for the light harvesting, but also the arrangement of the two kinds of organic dyes on the TiO2 surface.
DSCs based on the co-sensitization of LI-54/SQ2, LI-101/SQ2, LI-102/SQ2 and LI-103/SQ2 were fabricated with the same concentration of the single dye (0.3 mM). As shown in Fig. S2 and Table S1,† due to the co-sensitization approach, the conversion efficiency increased from 2.76% (SQ2) to 7.00% (LI-102/SQ2), together with the enhancement of Jsc and Voc in a large degree. With the aim to further optimize the photovoltaic performance of the co-sensitized solar cells, a low concentration of CDCA was added, an even higher conversion efficiency of 7.44% was achieved in the mixture of LI-102/SQ2 (Fig. 5 and Table 2). In comparison with the common dye LI-54 in the condition of co-sensitization, the advantage of the “H” structure was more obvious. The three “H” type dyes exhibited the conversion efficiencies increase by about 1.3 folds from 5.62% (LI-54/SQ2) to 7.10–7.44% (LI-101-LI-103/SQ2), mainly attributing to the increase of Voc. The co-sensitization of the “H” type dyes and SQ2 led to the enhancement of Voc from 632 mV (SQ2) to 685–718 mV (LI-101-LI-103/SQ2). However, for the LI-54/SQ2 co-sensitized system, the Voc value decreased to 620 mV, indicating that the “H” configuration was really helpful to suppressing the dye aggregates and electron recombination on the TiO2 surface.
Electrochemical impedance spectroscopy
To investigate the interfacial electron transition and recombination processes, the electrochemical impedance spectroscopy (EIS) was employed in DSCs in the dark under a forward bias of −0.70 V (frequency range: 10−1 to 105 Hz), to elucidate the relationship between the Voc and the molecular structure. Both the Nyquist and Bode plots of the DSCs were presented in Fig. 6. For the Nyquist plots, the semicircles in the middle frequency, which were related to the resistances of charge recombination (Rrec) at the interface of TiO2/electrolyte, were much different (Table 2). The bigger semicircle meant the larger Rrec, indicating the retardation of the interfacial charge recombination was more effectively. For the DSCs based on the single dyes, the values of Rrec increased in the order of LI-54 (55.13 Ω cm−2) < LI-101 (57.95 Ω cm−2) < LI-103 (78.26 Ω cm−2) < LI-102 (112.70 Ω cm−2), in good agreement with the trend of Voc values. The higher values of “H” type dyes indicated that the configuration of “H” structures could be effective to suppress the charge recombination for the more compact layer of sensitizers and the less ineffective interaction between the D–π–A moieties. Among the three “H” type dyes, LI-102 with meta-linkage mode exhibited the best performance, since the parallel configuration of the two D–π–A moieties would be beneficial to the alignment of the sensitizers on the TiO2 surface. After the co-sensitization with SQ2, the anti-aggregate property of the “H” type dyes were more obvious than that of the rod-sharp one (LI-54), the Rrec values and lifetimes (τ) of DSCs based on LI-101/SQ2, LI-102/SQ2 and LI-103/SQ2 were larger than that based on LI-54/SQ2, indicating that the severe aggregates and charge recombination in the DSC based on SQ2 (Rrec = 24.68 Ω cm−2, τ = 11.16 ms) could be suppressed by the “H” type dyes in a large degree. (LI-101–LI-103) more effectively than that of LI-54 with one D–π–A unit, and it was also supported by the enhancement of Voc values in the DSCs based on the co-sensitization of “H” type dyes and SQ2.
 |
| | Fig. 6 EIS spectra of DSCs tested at −0.7 V forward bias in the dark: (A) Nyquist plots and (B) Bode phase. | |
To further study the charge recombination kinetics of DSCs, the intensity-modulated photovoltage spectroscopy (IMVS) measurement was also employed.29,30 IMVS was performed under the incident light illumination, which resembled real working conditions of DSCs. Fig. 7 showed the electron lifetime obtained from IMVS measurement under different light intensities. For the DSCs based on the single dyes and co-sensitized systems, the trend of their electron lifetimes was in consistent with that of EIS data, disclosing that the smaller charge recombination contributed to the longer electron lifetime, resulting in the higher Voc values. It further confirmed that the combination of “H” type dyes and SQ2 was beneficial to optimizing the photovoltaic performance of DSCs.
 |
| | Fig. 7 Electron lifetime as a function of Voc for the DSCs on the single dyes and co-sensitized systems. | |
Conclusion
In summary, we have synthesized three H-type organic dyes LI-101–LI-103 by changing the linkage modes of the two D–π–A moieties, with the aim to optimize the configurations of “H” structures. After they were utilized for the co-sensitization with a NIR squaraine dye SQ2, the complementary absorption spectra of the H-type dyes and SQ2 can be beneficial to broadening the light-harvesting and improving Jsc values, and the anti-aggregated properties of “H” structures would contribute to achieving the regular alignment of the co-sensitizers on the TiO2 surface for the enhancement of Voc values. Thus, the LI-102/SQ2 co-sensitized solar cell exhibited conversion efficiency of 7.44%, with 16.6 mA cm−2 for Jsc, 714 mV for Voc, 0.63 for FF, much higher than that of LI-54 (similar structure with one D–π–A unit)/SQ2 (5.62%) and single SQ2 (4.28%).
Experimental section
Materials
Tetrahydrofuran (THF), N,N-dimethylformamide (DMF), and 1,2-dichloroethane were treated to remove the water and the other possible impurity according to the literatures.31 Phosphorus oxychloride (POCl3) was distilled before use. Compound 1 and diethyl 4-(diphenylamino)benzylphosphonate were synthesized as previously reported.32,33 All other reagents were purchased and used directly as received.
Instrumentation
1H NMR spectra were measured on a Varian Mercury 300 spectrometer by using tetramethylsilane (TMS; δ = 0 ppm) as internal standard. 13C NMR spectra were measured on a Bruker 700 spectrometer by using tetramethylsilane (TMS; δ = 0 ppm) as internal standard. MS (MALDI-TOF) spectra were recorded on a Waters Micromass LCT Premier XE. UV-visible spectra were measured on a Shimadzu UV-2550 spectrometer. Electrochemical cyclic voltammetry was conducted on a CHI 660 voltammetric analyzer with Pt disk, Ag/Ag+ electrode and Pt plate, as working, reference and counter electrodes, respectively, in anhydrous CH2Cl2 under an atmosphere of nitrogen, while the supporting electrolyte was tetrabutylammoniumhexafluorophosphate (TBAPF6) and a scanning rate was 100 mV s−1. Ferrocene/ferrocenium redox couple was used to conduct the potential calibration.
Synthesis of compound 2a
Under an atmosphere of nitrogen, K2CO3 (1.33 g, 9.60 mmol), compound 1 (2.06 g, 8.00 mmol) and pyrocatechol (0.35 g, 3.20 mmol) were added to acetone (20 mL) solution. The mixture was stirred at 75 °C for 60 h. After cooled to room temperature, the mixture was filtered, the filtrate was dried over and further purified by column chromatography on silica gel with the mixture of petroleum ether/ethyl acetate (6/1) as the eluent to yield compound 2a as a brown liquid (1.07 g, 71.7%). 1H NMR (CDCl3) δ (ppm): 9.52 (s, 2H, –CHO), 6.92 (s, br, 4H, ArH), 6.88 (s, br, 4H, ArH), 6.20 (m, 2H, ArH), 4.30 (t, J = 7.2 Hz, 4H, –N–CH2–), 3.96 (t, J = 6.9 Hz, 4H, –O–CH2–), 1.78 (m, 8H, –CH2–), 1.40 (m, 4H, –CH2–), 1.30 (m, 4H, –CH2–).
Synthesis of compound 2b
Compound 2b was synthesized according to the similar procedure of 2a as a brown liquid (1.22 g, 81.8%). 1H NMR (CDCl3) δ (ppm): 9.53 (s, 2H, –CHO), 6.93 (s, br, 4H, ArH), 6.80 (s, br, 4H, ArH), 6.21 (s, br, 2H, ArH), 4.31 (t, J = 7.1 Hz, 4H, –N–CH2–), 3.88 (t, J = 6.2 Hz, 4H, –O–CH2–), 1.74 (m, 8H, –CH2–), 1.48 (m, 4H, –CH2–), 1.35 (m, 4H, –CH2–).
Synthesis of compound 2c
Compound 2c was synthesized according to the similar procedure of 2a as a brown liquid (1.08 g, 72.7%). 1H NMR (CDCl3) δ (ppm): 9.53 (s, 2H, –CHO), 6.93 (s, br, 4H, ArH), 6.80 (s, br, 4H, ArH), 6.21 (s, br, 2H, ArH), 4.31 (t, J = 7.5 Hz, 4H, –N–CH2–), 3.88 (t, J = 6.0 Hz, 4H, –O–CH2–), 1.76 (m, 8H, –CH2–), 1.45 (m, 4H, –CH2–), 1.35 (m, 4H, –CH2–).
Synthesis of compound 3a
Under an atmosphere of nitrogen, potassium tert-butoxide (t-BuOK, 0.78 g, 6.88 mmol) in THF was added slowly into the THF solution (5 mL) of compound 2a (0.80 g, 1.72 mmol) and diethyl 4-(diphenylamino)benzylphosphonate in an ice-water bath. The mixture was stirred at 25 °C in dark for 12 h, then the pH value was adjusted to ∼7 with dilute hydrochloric acid. The resultant solution was extracted with chloroform, and dried over Na2SO4. The crude product was further purified by column chromatography on silica gel with the mixture solvent of petroleum ether/ethyl acetate (8:1) as eluent, to yield 3a as a yellow solid (1.50 g, 92.3%). 1H NMR (CDCl3) δ (ppm): 7.31–7.21 (m, 12H, ArH and –CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–), 7.08 (d, J = 7.8 Hz, 8H, ArH), 7.00 (m, 8H, ArH and –CHCH–), 6.82 (d, J = 4.2 Hz, 8H, ArH), 6.63 (s, br, 2H, ArH), 6.44 (s, br, 2H, ArH), 6.12 (s, br, 2H, ArH), 3.96–3.90 (m, 8H, –N–CH2– and –O–CH2–), 1.80–1.73 (m, 8H, –CH2–), 1.51–1.46 (m, 4H, –CH2–), 1.38–1.35 (m, 4H, –CH2–).
CH–), 7.08 (d, J = 7.8 Hz, 8H, ArH), 7.00 (m, 8H, ArH and –CHCH–), 6.82 (d, J = 4.2 Hz, 8H, ArH), 6.63 (s, br, 2H, ArH), 6.44 (s, br, 2H, ArH), 6.12 (s, br, 2H, ArH), 3.96–3.90 (m, 8H, –N–CH2– and –O–CH2–), 1.80–1.73 (m, 8H, –CH2–), 1.51–1.46 (m, 4H, –CH2–), 1.38–1.35 (m, 4H, –CH2–).
Synthesis of compound 3b
Compound 3b was synthesized according to the similar procedure of 3a as a yellow solid (700 mg, 90.9%). 1H NMR (CDCl3) δ (ppm): 7.32–7.21 (m, 12H, ArH and –CHCH–), 7.11–7.01 (m, 18H, ArH and –CHCH–), 6.82 (s, br, 4H, ArH), 6.65 (s, br, 2H, ArH), 6.43 (d, J = 8.4 Hz, 4H, ArH), 6.14 (s, br, 2H, ArH), 3.92 (m, 8H, –N–CH2– and –O–CH2–), 1.77 (s, br, 8H, –CH2–), 1.47–1.38 (m, 8H, –CH2–).
Synthesis of compound 3c
Compound 3c was synthesized according to the similar procedure of 3a as a yellow solid (991 mg, 60.8%). 1H NMR (CDCl3) δ (ppm): 7.32 (d, J = 8.4 Hz, 6H, ArH), 7.27–7.22 (m, 6H, ArH and –CHCH–), 7.10 (d, J = 7.5 Hz, 8H, ArH), 7.05–6.99 (m, 8H, ArH and –CHCH–), 6.83 (s, br, 4H, ArH), 6.77 (s, br, 4H, ArH), 6.65 (s, br, 2H, ArH), 6.45 (s, br, 2H, ArH), 6.15 (s, br, 2H, ArH), 3.96 (t, J = 6.9 Hz, 4H, –N–CH2–), 3.86 (t, J = 6.3 Hz, 4H, –O–CH2–), 1.80–1.71 (m, 8H, –CH2–), 1.52–1.37 (m, 8H, –CH2–).
Synthesis of compound 4a
Under an atmosphere of nitrogen, phosphorus oxychloride (322 mg, 2.10 mmol) was added to dry DMF (246 mg, 3.36 mmol) in an ice-water bath slowly with stirring to form a glassy solid. After 3a (800 mg, 0.84 mmol) in 1,2-dichloroethane (15 mL) was added, the mixture was stirred at 25 °C for 12 h. Then the pH value was adjusted to ∼7 with Na2CO3, and the resultant solution was extracted with chloroform, and dried over Na2SO4. The crude product was further purified by column chromatography on silica gel with the mixture solvent of petroleum ether/ethyl acetate (3:1) as eluent to yield 4a as a yellow solid (486 mg, 57.7%). 1H NMR (CDCl3) δ (ppm): 9.41 (s, 2H, –CHO), 7.32 (d, J = 8.7 Hz, 4H, ArH), 7.28–7.23 (m, 6H, ArH and –CHCH–), 7.10–6.98 (m, 20H, ArH and –CHCH–), 6.88 (d, J = 3.6 Hz, 2H, ArH), 6.84 (s, br, 2H, ArH), 6.80 (m, 4H, ArH), 6.51 (d, J = 4.2 Hz, 2H, ArH), 4.43 (t, J = 6.9 Hz, 4H, –N–CH2–), 3.92 (t, J = 6.3 Hz, 4H, –O–CH2–), 1.74 (m, 8H, –CH2–), 1.46–1.37 (m, 8H, –CH2–).
Synthesis of compound 4b
Compound 4b was synthesized according to the similar procedure of 4a as a yellow solid (262 mg, 41.5%). 1H NMR (CDCl3) δ (ppm): 9.44 (s, 2H, –CHO), 7.35 (d, J = 9.0 Hz, 4H, ArH), 7.29–7.24 (m, 6H, ArH), 7.12–7.02 (m, 21H, ArH and –CH–CH–), 6.91 (d, J = 4.2 Hz, 2H, ArH), 6.83 (d, J = 15.6 Hz, 2H, –CHCH–), 6.53 (d, J = 4.5 Hz, 2H, ArH), 6.41 (d, J = 7.8 Hz, 3H, ArH), 4.46 (t, J = 7.2 Hz, 4H, –N–CH2–), 3.88 (t, J = 6.6 Hz, 4H, –O–CH2–), 1.76 (s, br, 8H, –CH2–), 1.43 (s, br, 8H, –CH2–).
Synthesis of compound 4c
Compound 4c was synthesized according to the similar procedure of 4a as a yellow solid (506 mg, 60.0%).1H NMR (CDCl3) δ (ppm): 9.45 (s, 2H, –CHO), 7.35 (d, J = 8.7 Hz, 4H, ArH), 7.30–7.24 (m, 6H, ArH), 7.13–7.03 (m, 20H, ArH and –CHCH–), 6.91 (d, J = 4.2 Hz, 2H, ArH), 6.83 (d, J = 16.2 Hz, 2H, –CHCH–), 6.75 (s, br, 4H, ArH), 6.54 (d, J = 4.2 Hz, 2H, ArH), 4.47 (t, J = 7.5 Hz, 4H, –N–CH2–), 3.85 (t, J = 6.9 Hz, 4H, –O–CH2–), 1.74 (s, br, 8H, –CH2–), 1.42 (s, br, 8H, –CH2–).
Synthesis of sensitizer LI-101
Under an atmosphere of nitrogen, to a mixture of 5a (200 mg, 0.20 mmol), cyanoacetic acid (89 mg, 1.00 mmol) and piperidine (10 μL) was added acetonitrile (10 mL). Then the resultant solution was refluxed for 12 h. After cooled to room temperature, the solution was poured into water and extracted with CH2Cl2. The organic fractions were combined, washed with water and dried with Na2SO4. The crude product was further purified by column chromatography on silica gel with the mixture solvent of chloroform/methanol (50:1) as eluent to yield sensitizer LI-101 as a red solid (135 mg, 59.3%). 1H NMR (DMSO-d6) δ (ppm): 7.97 (s, 2H, –CH), 7.52 (d, J = 8.1 Hz, 6H, ArH), 7.33–7.28 (m, 10H, ArH and –CHCH–), 7.17 (d, J = 14.7 Hz, 2H, –CHCH–), 7.10–7.00 (m, 12H, ArH), 6.87 (d, J = 8.4 Hz, 6H, ArH), 6.80 (s, br, 2H, ArH), 6.76 (s, br, 2H, ArH), 4.23 (s, br, 4H, –N–CH2–), 3.80 (s, br, 4H, –O–CH2–), 1.57 (s, br, 8H, –CH2–), 1.39 (s, br, 4H, –CH2–), 1.27 (s, br, 4H, –CH2–). 13C NMR (THF-d8) δ (ppm): 164.8, 149.5, 148.1, 147.4, 141.0, 137.5, 132.3, 130.9, 129.2, 127.7, 124.6, 123.3, 122.8, 120.7, 119.4, 117.3, 114.0, 113.5, 110.1, 91.6, 68.5, 42.5, 31.9, 29.4, 26.2, 25.6. MS (ESI, m/z): [M − H]− calcd for C74H68N6O6 1135.5200; found 1135.5140.
Synthesis of sensitizer LI-102
Compound LI-102 was synthesized according to the similar procedure of LI-101 as a red solid (142 mg, 54.3%). 1H NMR (DMSO-d6) δ (ppm): 8.00 (s, 2H, –CH), 7.57 (d, J = 7.2 Hz, 6H, ArH), 7.33–7.26 (m, 10H, ArH and –CHCH–), 7.18 (d, J = 15.9 Hz, 2H, –CHCH–), 7.09–7.02 (m, 14H, ArH), 6.95–6.90 (m, 6H, ArH), 6.38 (d, J = 6.0 Hz, 2H, ArH), 4.30 (s, br, 4H, –N–CH2–), 3.84 (s, br, 4H, –O–CH2–), 1.61 (s, br, 8H, –CH2–), 1.35 (s, br, 8H, –CH2–). 13C NMR (THF-d8) δ (ppm): 164.7, 160.4, 148.1, 147.4, 141.0, 137.5, 131.4, 130.9, 129.4, 129.1, 127.8, 127.6, 124.6, 123.3, 122.8, 119.4, 117.3, 113.6, 110.1, 101.1, 91.6, 78.5, 67.3, 42.5, 33.8, 31.6, 26.3, 25.7. MS (ESI, m/z): [M − H]− calcd for C74H68N6O6 1135.5200; found 1135.5153.
Synthesis of sensitizer LI-103
Compound LI-103 was synthesized according to the similar procedure of LI-101 as a red solid (150 mg, 65.9%). 1H NMR (DMSO-d6) δ (ppm): 8.01 (s, 2H, –CH), 7.58 (m, 6H, ArH), 7.33–7.28 (m, 10H, ArH and –CHCH–), 7.20 (d, J = 16.5 Hz, 2H, –CHCH–), 7.09–7.02 (m, 12H, ArH), 6.96 (d, J = 4.5 Hz, 2H, ArH), 6.92 (d, J = 8.4 Hz, 6H, ArH), 6.70 (s, br, 4H, ArH), 4.30 (s, br, 4H, –N–CH2–), 3.78 (s, br, 4H, –O–CH2–), 1.60 (s, br, 8H, –CH2–), 1.38–1.23 (m, 8H, –CH2–). 13C NMR (THF-d8) δ (ppm): 164.8, 155.2, 153.2, 148.1, 147.4, 141.1, 137.6, 132.3, 130.9, 129.3, 126.2, 124.6, 124.4, 123.3, 122.6, 119.4, 114.9, 113.6, 91.6, 71.4, 44.0, 31.6, 29.3, 26.3, 25.7. MS (ESI, m/z): [M − H]− calcd for C74H68N6O6 1135.5200; found 1135.5166.
Fabrication of solar cells
The dye-sensitized TiO2 electrode was prepared as reported in the literature.34 The FTO conductive glass was washed with water, acetone and ethanol. Then the cleaned conducting glass substrates (FTO, 2.2 mm thickness, 7–8 ohm sq−1) were immersed in a solution of TiCl4 (40 mM) at 70 °C for 30 min. After they were cooled to room temperature, washed with water and ethanol. The FTO conducting glass was coated by a layer (ca. 4 μm) of TiO2 paste (18NR-T, Dyesol) through screen printing and then dried for 6 min at 125 °C. This process was repeated four times to give a transparent film. Finally, a 4 μm scatter layer (18NR-AO, Dyesol) was coated to yield the final film with a thickness of about 16 μm. Then the TiO2 electrodes were gradually heated under airflow at 325 °C for 5 min, 375 °C for 5 min, 450 °C for 15 min and 500 °C for 1 h. The film were treated by a solution of TiCl4 (40 mM) at 70 °C for 30 min again after cooled to room temperature, then washed with water, ethanol and dried. Lastly, the TiO2 electrodes were heated at 500 °C for 30 min. After final sintering, the TiO2 electrodes was cooled to room temperature and immersed in a dye solution for 24 h. Solutions of LI-54, LI-101, LI-102, LI-103, SQ2, LI-54/SQ2 (1/1), LI-101/SQ2 (1/1), LI-102/SQ2 (1/1) and LI-103/SQ2 (1/1) (3 × 10−4 M) were prepared with the solvent system of acetonitrile and t-BuOH (1/1) at room temperature. The counter electrode was made by a conducting glass substrate (FTO, 2.2 mm thickness, 7–8 ohm sq−1) with spreading a 20 mM solution of H2PtCl6 in isopropyl alcohol, which was drilled a small hole to allow the introduction of the liquid electrolyte under vacuum. The counter electrode heated at 400 °C for 30 min before use. The sensitized electrodes were washed with corresponding solvent and dried under air, then the adsorbed TiO2 electrode and Pt counter electrode were assembled into a sealed sandwich-type cell by heating with a hot-melt gasket of 25 μm thickness (the ionomer Surlyn 1702, DuPont). The electrolyte was placed in the hole and sealed by using a piece of aluminum foil tape. The electrolyte was composed of 0.6 M dimethylpropylimidazolium iodide, 0.1 M lithium iodide, 0.03 M iodine, 0.5 M tert-butylpyridine in acetonitrile (ACN)/3-methoxypropionitrile (MPN) (1:1, v/v).
Photovoltaic properties measurements
The DSC was illuminated by light with energy of a 100 mW cm−2 from 450 W AM 1.5G simulated sunlight (Model 94023A, Newport Co.). The light intensity was determined using a Si solar cell (Model 91150, Newport Co.) as reference. The current–voltage (J–V) curves for the fabricated DSCs were obtained by applying a Keithley model 2400 digital source meter. Incident photon-current conversion efficiency (IPCE) was recorded on a DC Power Meter (Model 2931-C, Newport Co.) under irradiation of a 300 W xenon lamp light source with a motorized monochromator (Oriel). The xenon lamp was powered by an Arc Lamp Power Supply (Model 69920, Newport Co.). The electrochemical impedance spectroscopy (EIS) was measured with a frequency range of 0.1 Hz to 100 kHz in dark under a forward bias of −0.70 V on a CHI660E electrochemical workstation. The intensity modulated photovoltage spectroscopy (IMVS) was performed by a Solartron Analytical Modulab DSC with a monochromatic light source (590 nm) using a previously reported method.
Acknowledgements
We are grateful to the National Science Foundation of China (no. 21372003, and 21325416) for financial support.
Notes and references
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 CrossRef CAS PubMed.
- B. V. S. R. Umer Mehmood, S.-U. Rahman, K. Harrabi and I. A. Hussein, Adv. Mater. Sci. Eng., 2014, 2014, 1–12 CrossRef.
- X. Xie, K. Kretschmer and G. Wang, Nanoscale, 2015, 7, 13278–13292 RSC.
- T. Higashino, Y. Fujimori, K. Sugiura, Y. Tsuji, S. Ito and H. Imahori, Angew. Chem., Int. Ed., 2015, 54, 9052–9056 CrossRef CAS PubMed.
- J. Liu, B. Liu, Y. Tang, W. Zhang, W. Wu, Y. Xie and W.-H. Zhu, J. Mater. Chem. C, 2015, 3, 11144–11150 RSC.
- Z. Chai, M. Wu, M. Fang, S. Wan, T. Xu, R. Tang, Y. Xie, A. Mei, H. Han, Q. Li and Z. Li, Adv. Energy Mater., 2015, 5, 1500846 Search PubMed.
- K. Pei, Y. Wu, A. Islam, Q. Zhang, L. Han, H. Tian and W. Zhu, ACS Appl. Mater. Interfaces, 2013, 5, 4986–4995 CAS.
- Z. Yao, M. Zhang, R. Li, L. Yang, Y. Qiao and P. Wang, Angew. Chem., Int. Ed., 2015, 54, 5994–5998 CrossRef CAS PubMed.
- J.-S. Ni, Y.-C. Yen and J. T. Lin, Chem. Commun., 2015, 51, 17080–17083 RSC.
- Y. Wu, M. Marszalek, S. M. Zakeeruddin, Q. Zhang, H. Tian, M. Gratzel and W. Zhu, Energy Environ. Sci., 2012, 5, 8261–8272 CAS.
- M. Liang and J. Chen, Chem. Soc. Rev., 2013, 42, 3453–3488 RSC.
- S. Ahmad, E. Guillen, L. Kavan, M. Gratzel and M. K. Nazeeruddin, Energy Environ. Sci., 2013, 6, 3439–3466 CAS.
- K. Sayama, S. Tsukagoshi, T. Mori, K. Hara, Y. Ohga, A. Shinpou, Y. Abe, S. Suga and H. Arakawa, Sol. Energy Mater. Sol. Cells, 2003, 80, 47–71 CrossRef CAS.
- M. Guo, P. Diao, Y. J. Ren, F. Meng, H. Tian and S. M. Cai, Sol. Energy Mater. Sol. Cells, 2005, 88, 23–35 CrossRef CAS.
- Y. Chen, Z. Zeng, C. Li, W. Wang, X. Wang and B. Zhang, New J. Chem., 2005, 29, 773–776 RSC.
- T. Geiger, S. Kuster, J. H. Yum, S. J. Moon, M. K. Nazeeruddin, M. Grätzel and F. Nüesch, Adv. Funct. Mater., 2009, 19, 2720–2727 CrossRef CAS.
- R. Y. Y. Lin, Y. S. Yen, Y. T. Cheng, C. P. Lee, Y. C. Hsu, H. H. Chou, C. Y. Hsu, Y. C. Chen, J. T. Lin, K. C. Ho and C. Tsai, Org. Lett., 2012, 14, 3612–3615 CrossRef CAS PubMed.
- Z. Xue, L. Wang and B. Liu, Nanoscale, 2013, 5, 2269–2273 RSC.
- L. Y. Lin, M. H. Yeh, C.
P. Lee, J. Chang, A. Baheti, R. Vittal, K. R. Justin Thomas and K. C. Ho, J. Power Sources, 2014, 247, 906–914 CrossRef CAS.
- A. Connell, P. J. Holliman, M. L. Davies, C. D. Gwenin, S. Weiss, M. B. Pitak, P. N. Horton, S. J. Coles and G. Cooke, J. Mater. Chem. A, 2014, 2, 4055–4066 CAS.
- Q. Li, J. Shi, H. Li, S. Li, C. Zhong, F. Guo, M. Peng, J. Hua, J. Qin and Z. Li, J. Mater. Chem., 2012, 22, 6689–6696 RSC.
- K. D. Seo, B. S. You, I. T. Choi, M. J. Ju, M. You, H. S. Kang and H. K. Kim, Dyes Pigm., 2013, 99, 599–606 CrossRef CAS.
- Z. Ning, Y. Zhou, Q. Zhang, D. Ma, J. Zhang and H. Tian, J. Photochem. Photobiol., A, 2007, 192, 8–16 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian, Inc., Wallingford CT, 2009.
- K. D. Seo, B. S. You, I. T. Choi, M. J. Ju, M. You, H. S. Kang and H. K. Kim, J. Mater. Chem. A, 2013, 1, 9947–9953 CAS.
- K. D. Seo, B. S. You, I. T. Choi, M. J. Ju, M. You, H. S. Kang and H. K. Kim, ChemSusChem, 2013, 6, 2069–2073 CrossRef CAS PubMed.
- J. Li, W. Wu, J. Yang, J. Tang, Y. Long and J. Hua, Sci. China: Chem., 2011, 54, 699–706 CAS.
- J. Shi, Z. Chai, R. Tang, J. Hua, Q. Li and Z. Li, Sci. China: Chem., 2015, 58, 1144–1151 CrossRef CAS.
- J. Bisquert, D. Cahen, G. Hodes, S. Rühle and A. Zaban, J. Phys. Chem. B, 2004, 108, 8106–8118 CrossRef CAS.
- T. Marinado, K. Nonomura, J. Nissfolk, M. K. Karlsson, D. P. Hagberg, L. Sun, S. Mori and A. Hagfeldt, Langmuir, 2010, 26, 2592–2598 CrossRef CAS PubMed.
- H. Li, Y. Hou, Y. Yang, R. Tang, J. Chen, H. Wang, H. Han, T. Peng, Q. Li and Z. Li, ACS Appl. Mater. Interfaces, 2013, 5, 12469–12477 CAS.
- M. I. L. Soares, S. M. M. Lopes, P. F. Cruz, R. M. M. Brito and T. M. V. D. Pinho e Melo, Tetrahedron, 2008, 64, 9745–9753 CrossRef CAS.
- S. Zheng, S. Barlow, T. C. Parker and S. R. Marder, Tetrahedron Lett., 2003, 44, 7989–7992 CrossRef CAS.
- S. Ito, T. N. Murakami, P. Comte, P. Liska, C. Grätzel, M. K. Nazeeruddin and M. Grätzel, Thin Solid Films, 2008, 516, 4613–4619 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Chart S1, Fig. S1, S2 and Table S1. See DOI: 10.1039/c6ra03694e |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 








