
Surface enhanced Raman scattering based reaction monitoring of in vitro decyclization of creatinine → creatine†
Abstract
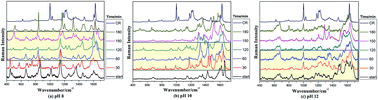
Creatinine → creatine decyclization is an important reaction wherein we obtain a beneficial compound from a toxic substance. Decyclization of creatinine at basic pH has been monitored in vitro by time series surface enhanced Raman scattering (SERS) using a silver island film. NH2 scissoring, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N and CO stretching modes of creatinine serve as Raman markers for monitoring the decyclization reaction. Transition state calculations using DFT have revealed the path of the formation of creatine by the cleavage of the endocyclic CN bond of creatinine. The Raman signatures of ring opening are clearly observed after 120 min at pH 8, and further increasing the pH increases the reaction rate even more, as the signatures are observed after 60 and 30 min at pH 10 and 12, respectively; however, the reversibility of the reaction is more prominent at higher pH. Therefore, pH 8 is the most favorable among the three pH values for the decyclization reaction to be stable at room temperature. The proper understanding of this reaction where a toxic substance is converted to a beneficial compound is expected to open the scope for further extensive research.
N and CO stretching modes of creatinine serve as Raman markers for monitoring the decyclization reaction. Transition state calculations using DFT have revealed the path of the formation of creatine by the cleavage of the endocyclic CN bond of creatinine. The Raman signatures of ring opening are clearly observed after 120 min at pH 8, and further increasing the pH increases the reaction rate even more, as the signatures are observed after 60 and 30 min at pH 10 and 12, respectively; however, the reversibility of the reaction is more prominent at higher pH. Therefore, pH 8 is the most favorable among the three pH values for the decyclization reaction to be stable at room temperature. The proper understanding of this reaction where a toxic substance is converted to a beneficial compound is expected to open the scope for further extensive research.
 Please wait while we load your content...
Please wait while we load your content...

