
Inactivation of immobilized trypsin under dissimilar conditions produces trypsin molecules with different structures†
Alfredo Sanchez‡
a,
Jenifer Cruz‡bc,
Nazzoly Ruedabc,
Jose C. S. dos Santosbd,
Rodrigo Torresce,
Claudia Ortizf,
Reynaldo Villalonga*a and
Roberto Fernandez-Lafuente*b
aDepartment of Analytical Chemistry, Faculty of Chemistry, Complutense University of Madrid, 28040 Madrid, Spain. E-mail: rvillalonga@quim.ucm.es
bDepartamento de Biocatálisis, Instituto de Catálisis-CSIC, C/Marie Curie 2, Campus UAM-CSIC, Cantoblanco, 28049 Madrid, Spain. E-mail: rfl@icp.csic.es
cEscuela de Química, Grupo de investigación en Bioquímica y Microbiología (GIBIM), Edificio Camilo Torres 210, Universidad Industrial de Santander, Bucaramanga, Colombia
dInstituto de Engenharias e Desenvolvimento Sustentável, Universidade da Integração Internacional da Lusofonia Afro-Brasileira, CEP 62785-000, Acarape, CE, Brazil
eCurrent address: Laboratorio de Biotecnología, Instituto Colombiano del Petróleo-Ecopetrol, Piedecuesta, Bucaramanga, Colombia
fEscuela de Microbiología, Universidad Industrial de Santander, Bucaramanga, Colombia
First published on 10th March 2016
Abstract
Bovine trypsin has been immobilized on glyoxyl-agarose and two different preparations have been produced. One was reduced just after immobilization, while the other was left to continue the enzyme-support reaction. This strategy is a guarantee of the identical orientation of the enzyme regarding the support surface and the identical physical properties of the support. Then, the two preparations were submitted to inactivations under different conditions: thermal and solvent inactivations under different pH values. After drying, the structures of the different enzymes preparations were analyzed by deconvolution of the amide I region, which provides information about the secondary structure of the protein in terms of α-helixes, β-sheets, β-turns and non-ordered or irregular structures. The results confirm that the structures of the different preparations were very different, suggesting that the inactivation ways were different for each enzyme preparation and dependent on the inactivation conditions. This information is very relevant for the design of strategies for enzyme stabilization, as shown by the fact that the inactivation may follow different conformational changes depending on the degree of enzyme rigidification and inactivation conditions.
1. Introduction
Enzymes are very interesting biocatalysts, their high activity under mild conditions, together with their high specificity and selectivity makes them a very good alternative to conventional chemistry or catalysis for complex or labile compounds, and may be a good alternative for very contaminant processes.1–4However, enzymes have a biological origin and thus some properties may not really fit the industrial requirements.5 Among these properties, moderate enzyme stability, necessary in vivo to permit a rapid answer to changes in the environment, becomes one of the greater problems when they are used as industrial catalysts.6–8 Enzyme inactivation of a monomeric enzyme starts by some reversible conformational changes, and finally the enzyme may also suffer some chemical modifications, aggregation, etc.9–12 That way, most strategies to stabilize enzymes are directed to the slowdown of these initial conformational changes. Enzyme stability has been improved by genetic tools (e.g., site-directed mutagenesis,13,14 chemical modification15–17 or immobilization18,19). Directed evolution is another very efficient technique to stabilize enzymes, the in vitro selection of the stabilized enzymes may be already performed under the desired conditions.20,21 Immobilization improves enzyme stability if several enzyme subunits (in multimeric proteins) are involved in the immobilization,22 or if several enzyme-support bonds are established increasing enzyme rigidity.22,23 This last fact increases the enzyme global rigidity and that way reduces the conformational changes, sometimes without reductions on enzyme activity. Considering that immobilization is required to facilitate enzyme recovery and reuse in many of the industrial uses of enzymes as biocatalysts,19,24–39 a great effort has been paid to couple immobilization to the solution of other enzyme limitations, no reducing this effector to the improvement of the enzyme stability, but also to tune enzyme selectivity, specificity or resistance to inhibitors18–29 or enzyme purification.30
It is assumed that the conformational changes start by some weak point of the enzyme conformation and then are getting more generalized along the whole enzyme conformation until reaching full enzyme inactivation.31–34 However, it is not difficult to imagine that the weakest point of an enzyme conformation, or at least the way that the enzyme structure follows during inactivation, may be different under different inactivating conditions.
It is remarkable that the stabilization of an enzyme via multipoint covalent attachment tends to be very different when evaluated under different inactivating conditions, and a immobilization via a region is critical for some inactivation conditions and not so relevant for others (e.g., as presented in the interesting paper from Grazu et al. using site directed rigidification35,36). Multipoint covalent attachment permits to increase the overall enzyme rigidity, but that does not occur with the same intensity in all the enzyme structure. The immobilized enzyme will have some conformational movements slower than others and that may produce that the new more rapid conformational change may be affecting a different area when compared to a non-stabilized enzyme. Moreover, the multipoint covalent attachment may produce some conformational changes, facilitating the generation of new ways for further enzyme conformational changes. Fig. 1 show this hypothesis.
 | ||
| Fig. 1 Scheme of the different conformational changes suffered by the same enzyme immobilized preparation under different conditions. | ||
Using free enzymes the study of the inactivation pathway may be complex, because the partially unfolded protein with tend to precipitate and that make the individual inactivation changes hard to follow. Moreover, from an applied point of view, the effects of immobilization and inactivation conditions on enzyme inactivation may be more interesting.
In this sense, it is not easy to measure the conformation of an enzyme in a solid state.37 One approximation to get this objective is the use of the infrared spectra of the proteins, in particular the amide I region at 1700–1600 cm−1, which is the major absorption band in proteins; this band is mostly originated by the stretching vibrations of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–N bonds.38 This band has been analyzed by deconvolution of the amide I region, that provides information about the secondary structure of the protein in terms of α-helix, β-sheets, β-turns and non-ordered or irregular structures.39–43 The studies involve the drying of the immobilized enzyme, and that may alter the enzyme structure due to the promotion of interactions between enzyme and support. However, if the support surface and enzyme orientation is exactly the same, the differences between different treated immobilized enzymes should be related only to different forms before this drying. Furthermore, if the support surface is very inert, this problem may be at least partially solved.44
O and C–N bonds.38 This band has been analyzed by deconvolution of the amide I region, that provides information about the secondary structure of the protein in terms of α-helix, β-sheets, β-turns and non-ordered or irregular structures.39–43 The studies involve the drying of the immobilized enzyme, and that may alter the enzyme structure due to the promotion of interactions between enzyme and support. However, if the support surface and enzyme orientation is exactly the same, the differences between different treated immobilized enzymes should be related only to different forms before this drying. Furthermore, if the support surface is very inert, this problem may be at least partially solved.44
Thus, this paper shows a study on the structural changes of trypsin immobilized on glyoxyl support via limited attachment or multipoint covalent attachment after inactivation under different conditions. Glyoxyl-agarose was selected a support because it is very inert after reduction45 and also because it produces an immobilization via a fixed area, the richest Lys containing surface area.46 By controlling the immobilization time, it is possible to alter the extent of the enzyme-support multipoint attachment,47 ensuring the exact identical orientation of the enzyme in both derivatives regarding the support surface (Fig. 2). Trypsin immobilized on this support has shown to maintain almost full activity versus macromolecular substrates48–50 and also to be fully inhibited by large trypsin inhibitors51,52, confirming a quite homogenous orientation of the enzyme that produces a biocatalyst with the active center fully exposed to the reaction media.
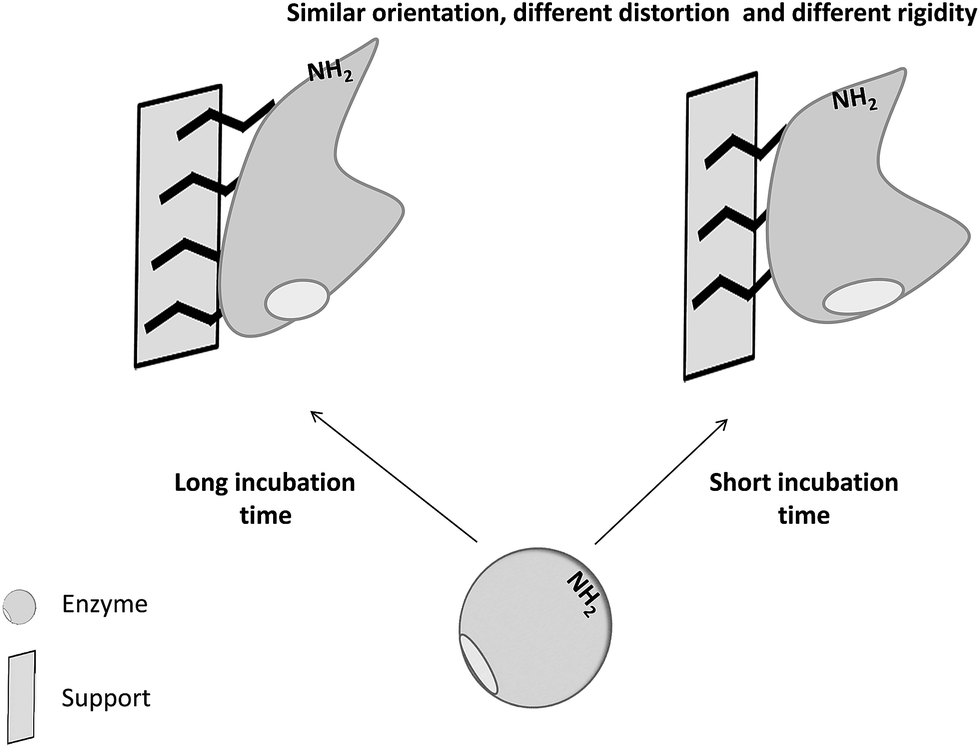 | ||
| Fig. 2 Scheme of the different enzyme structures of an immobilized enzyme when increasing the enzyme-support multi-interaction. | ||
2. Materials and methods
2.1. Materials
Bovine trypsin (E.C. 3.4.21.4), benzoyl-arginine p-nitroanilide (BANA), ethylenediamine (EDA), benzamidine, were purchased from Sigma Chemical Co. (St. Louis, MO, USA). Agarose beads 4BCL support were purchased from Agarose Bead Technologies (ABT), Spain. All other reagents were of analytical grade. Fully activated glyoxyl support was prepared as previously described.53All experiments were performed using three independent samples and the results are reported as the mean of these values and the standard deviation (usually under 10%).
2.2. Enzyme immobilization
A 10 g portion of support was suspended in 100 mL of trypsin solution (10 mg protein per g support) in 50 mM sodium carbonate at pH 10 and 25 °C containing 3 mM benzamidine. Just after immobilization, a fraction of the immobilized enzyme was separated and reduced by adding solid NaBH4 (final concentration of 1 mg ml−1)47 leaving this suspension for 1 hour under gentle stirring at room temperature, and then washed with abundant distilled water to eliminate residual sodium borohydride (derivative 1). The other portion of the immobilized enzyme was left to react with the support for 90 h before the reduction step (derivative 2). This second preparation has more time to react with the support and is expected to have a more intense multipoint attachment than the first one, and also permitted to have a higher degree of enzyme distortion due to the reaction with the support, the number of enzyme-support bonds has been quantified previously.472.3. Enzymatic assays
The activity of the soluble or immobilized enzyme was assayed by determination of the increase in absorbance at 405 nm which accompanies the hydrolysis of the synthetic substrate BANA. 100 or 200 microlitres of soluble or suspended enzyme were added to 2.5 mL of 50 mM sodium phosphate containing 30% (v/v) ethanol at pH 7 having 2 mM BANA, at 25 °C.282.4. Enzyme inactivations
Both immobilized trypsin preparations were submitted to identical inactivation conditions until the activity of the immobilized enzyme using the BANA assay described above decreased to 20%. The enzymes were incubated at pH 5 and 7 at 80 °C (in 50 mm sodium acetate or sodium phosphate respectively) or at 60 °C at pH 9 (in 50 mM sodium borate). In the inactivations in 80% (v/v) dioxane was performed at pH 5 and 9 and 60 °C, using 100 mM Tris buffer.2.5. Secondary structure studies of the immobilized enzymes
The immobilized enzyme preparations, submitted to different previous treatments and incubated for at least one week in 25 mM sodium phosphate at pH 7 and 4 °C after these treatments, were washed with distilled water, and dried at room temperature under ambient atmosphere. The secondary structures for all immobilized enzyme preparations, before and after different inactivation treatments, were evaluated according to amide I bands (1700–1600 cm−1) in the infrared spectra. The analyses were developed by FT-ATR-IR spectroscopy using an Alpha-T FTIR spectrometer (Bruker) with a resolution of 1.5 cm−1. The secondary structure contents were calculated through the areas obtained for the different bands and its fraction related to the whole amide I band. These areas were quantified after Gaussian–Newton deconvolution of the spectra. The analyzed bands and their assignment structures are detailed in Table 1.| Band position (cm−1) | Assignment |
|---|---|
| 1628 | β-Sheet |
| 1636 | β-Sheet |
| 1647 | Unordered |
| 1656 | α-Helix |
| 1667 | β-Turns |
| 1682 | Unordered |
3. Results & discussion
3.1. Enzyme immobilization
Fig. 3 shows the very rapid immobilization observed using glyoxyl agarose under the described conditions. The catalytic activity obtained for the enzyme immobilized after just 1 h was almost 100% of the offered one, due to the protecting effect of benzamidine during the enzyme-support reaction.47,54 After 90 h of enzyme support reaction, the recovered activity was still over 90%, not being possible to ensure a decrease on the enzyme activity due to the multi-interaction with the support. These data were obtained using a preparation with only 1 mg g−1 of derivative to ensure the absence of diffusional problems. | ||
| Fig. 3 Immobilization course of trypsin in glyoxyl-agarose. Conditions are described in methods section. Squares: activity in the supernatant of the immobilization suspension. Triangles: free enzyme under identical conditions. | ||
3.2. Characterization the secondary structure of both preparations
It should be remarked that these studies have been performed on enzymes immobilized on identical supports and conditions, using identical loading and distribution on the enzyme pores, and one of them is just the prolongation of the incubation time before reduction of the other preparation after full enzyme immobilization. This guarantees that both enzymes have identical enzyme orientations and may suffer identical interactions with the support during sample preparation. Thus, differences should be the result of real different enzyme structures produced by the longer incubation time. Infrared spectra are shown in Fig. 1-S.†Table 2 shows that the structures of both immobilized enzymes, even having very similar expressed activities, have some differences: derivative 2 showed a significant decrease in β-sheet content and a very clear increment on α-helix, while β-turns and unordered structures were less affected.
| Biocatalyst | Inactivation conditions | Time (min) | Residual activity (%) |
|---|---|---|---|
| 1 | pH 5 | 1200 ± 15.2 | 19.7 |
| pH 5-diox | 48 ± 3.4 | 21.3 | |
| pH 7 | 720 ± 2.1 | 22 | |
| pH 9 | 720 ± 6.2 | 18.4 | |
| pH 9-diox | 120 ± 7.6 | 21.6 | |
| 2 | pH 5 | 1420 ± 24.5 | 23.3 |
| pH 5-diox | 600 ± 5.6 | 19.9 | |
| pH 7 | 1080 ± 3.5 | 20.5 | |
| pH 9 | 840 ± 8.2 | 22.8 | |
| pH 9-diox | 360 ± 2.3 | 23 |
3.3. Enzyme inactivation under different conditions of immobilized preparations 1 and 2 of trypsin
Both enzyme preparations were submitted to the conditions described in methods section to achieve the enzyme inactivation until reaching a 20% of residual activity. Table 3 shows the time required for each preparation to reach this value under each specified condition. As expected, preparation 2 always required longer incubation time-periods to reach 20% of residual activity, although differences depend on the inactivating conditions. In 80% dioxane at pH 9, three folds longer time is required for preparation 2 than for preparation 1 to reach the desired activity value. At 80 °C in aqueous medium, this difference is hardly a 10%.| Biocatalyst | β-Sheet area (%) | α-Helix area (%) | β-Turns area (%) | Unordered area (%) |
|---|---|---|---|---|
| 1 | 68.3 ± 2 | 2.9 ± 0.08 | 6.7 ± 0.1 | 22.1 ± 1.2 |
| 2 | 59.7 ± 1 | 11.6 ± 0.9 | 9.3 ± 0.4 | 19.3 ± 0.9 |
It should be stated that agarose 4BCLm is not very suitable to give a very intense multipoint attachment due to the diameter of the trunks forming the agarose,47 and that the first enzyme immobilization involves as minimum 2 groups of the enzyme.46 Therefore, the differences in stability between both preparations are significant but not extremely high.
3.4. Changes induced immobilized trypsin after suffering inactivations under different conditions
Original infrared spectra are shown in Fig. 1-S.†Table 4 shows some clear differences on the changes of the secondary structure when preparation 1 is submitted to different inactivation conditions. It should be remarked that the enzyme preparations have been incubated under identical and mild conditions for at least one week before performing the experiments of structure determination, therefore the changes are not due to different conditions during the treatment for activity determination.
| Inactivation conditions | β-Sheet area (%) | α-Helix area (%) | β-Turns area (%) | Unordered area (%) |
|---|---|---|---|---|
| None | 1.0 | 1.0 | 1.0 | 1.0 |
| pH 5 | 1.1 ± 0.08 | 1.0 ± 0.03 | 1.0 ± 0.02 | 0.8 ± 0.02 |
| pH 5-diox | 1.1 ± 0.07 | 1.2 ± 0.06 | 1.2 ± 0.07 | 0.6 ± 0.02 |
| pH 7 | 0.7 ± 0.02 | 2.8 ± 0.1 | 0.8 ± 0.05 | 1.8 ± 0.1 |
| pH 9 | 0.6 ± 0.01 | 4.8 ± 0.25 | 3.5 ± 0.25 | 0.9 ± 0.03 |
| pH 9-diox | 0.4 ± 0.01 | 7.4 ± 0.3 | 2.6 ± 0.08 | 1.5 ± 0.1 |
Thermal inactivation at pH 5 of this preparation produced, as most significant changes, an increase in β-sheet content (almost a 10%), and a decrease of the unordered regions (almost a 40%). On the contrary, a significant increase of unordered structure and a very significant decrement of β-sheet were observed after incubation at pH 7.0. In addition, a noticeable decrease in the β-turn percentage was observed. Thermal inactivation at pH 9 also produced a completely different picture, being the most relevant changes an increase of β-turns and α-helix and a decrease of β-sheet, while the unordered structure remains under similar values.
If the inactivation of preparation 1 is performed in the presence of organic dioxane, the most relevant change is a decrease of the percentage of unordered structure at pH 5, while all other structures increased. In inactivations at pH 9, β-turns and α-helix content increased while that of β-sheet and unordered structure decreased.
The very different results show that this preparation reached very different enzyme conformations after experiencing different inactivation treatment, suggesting that the ways of enzyme inactivation may be different for each inactivation conditions.
Table 5 shows a similar study using the preparation 2. In aqueous medium, incubation at pH 5 of preparation 2 caused an increase in the β-sheet and α-helix content while the content β-turns and unordered structure decreased. At pH 7, the most relevant changes are an increase of β-sheet and a decrease of β-turns, while at pH 9 the increase of β-sheet is much more significant and the other structures decreased. In the presence of dioxane, again the changes are significant and very different. At pH 5, β-sheet and unordered content slightly increased while β-turns and α-helix significantly decreased, while at pH 9 in the presence of dioxane the changes are very similar to the result obtained in aqueous buffer, with a very significant increment of β-sheet. The results suggested that the structure of the inactive enzyme at pH 9 is similar, both in dioxane or aqueous media, while at pH 5 the changes in the structure are very different in both media, and also at pH 7 the changes are different to found at pH 5 or 9. Again, except for the inactivation at pH 9 in both media, different ways of enzyme inactivation when the enzyme is submitted to different inactivation conditions may be suggested from the results.
| Sample | β-Sheet area (%) | α-Helix area (%) | β-Turns area (%) | Unordered area (%) |
|---|---|---|---|---|
| None | 1.0 | 1.0 | 1.0 | 1.0 |
| pH 5 | 1.1 ± 0.08 | 1.3 ± 0.07 | 0.3 ± 0.01 | 0.7 ± 0.01 |
| pH 5-diox | 1.0 ± 0.06 | 0.4 ± 0.02 | 1.0 ± 0.03 | 1.2 ± 0.05 |
| pH 7 | 1.1 ± 0.04 | 0.9 ± 0.05 | 0.2 ± 0.01 | 1.0 ± 0.04 |
| pH 9 | 1.3 ± 0.05 | 0.3 ± 0.01 | 0.6 ± 0.02 | 0.7 ± 0.03 |
| pH 9-diox | 1.3 ± 0.08 | 0.4 ± 0.01 | 0.5 ± 0.01 | 0.7 ± 0.04 |
3.5. Comparison of the changes suffered by both enzyme preparations under identical inactivation conditions
Tables 4 and 5 show that the changes suffered for each preparation were quite different. For example, in aqueous media at pH 5 and 80 °C preparation 1 decrease its unordered structure percentage while preparation 2 mainly increased the percentage of α-helix. At pH 7, preparation 1 decreased β-sheet while preparation 2 increased it. At pH 9, preparation 1 increased α-helix and decreased β-sheet, while preparation 2 shows the opposite changes. In organic solvents the differences in the changes are also relevant. Thus, it is possible to state that the different rigidified enzyme preparations, even after being submitted to similar inactivation protocols until reaching similar activity recoveries, suffer very different conformational changes. This suggests that the rigidification may not only slow down the inactivation rate, but to change the area that suffer the most rapid conformational changes (e.g., by making more rigid the area affected on the less stabilized enzyme). Together to multipoint covalent attachment to preexisting solids,18,19 a recent emerging enzyme immobilization strategy is encapsulating enzyme molecules inside inorganic crystals which are considered as “hard” matter. Enzyme encapsulated in these inorganic crystals exhibited greatly increased stability due the fixed and rigid structure.55 Thus, many experimental studies have shown that increased rigidity of protein can highly improve enzyme stability at harsh conditions, and the results shown in this paper may explain some of the results.4. Conclusion
We have been able to show, using the deconvolution of the amide I region FT-ATR-IR spectra, that enzymes inactivated under different conditions suffer different conformational changes, suggesting that the inactivation areas involved in each condition may be different depending on the condition. Similarly, the changes induced on preparations with different rigidity may be quite different, suggesting that the inactivation may follow different ways depending on the rigidification of the different areas. This may open new ways to the development of stabilized preparations of immobilized enzymes.Acknowledgements
We gratefully recognize the support from the MINECO of Spanish Government, by the grant CTQ2013-41507-R and project CTQ2014-58989-P. The fellowships for Miss Cruz, Miss Rueda, (Colciencias) and Dr dos Santos (CNPq, Brazil) are also recognized. The help and comments from Dr Ángel Berenguer (Instituto de Materiales, Universidad de Alicante) are kindly acknowledged.References
- S. Jemli, D. Ayadi-Zouari, H. B. Hlima and S. Bejar, Crit. Rev. Biotechnol., 2016, 36, 246–258 CrossRef CAS PubMed.
- N. Tibrewal and Y. Tang, Annu. Rev. Chem. Biomol. Eng., 2014, 5, 347–366 CrossRef CAS PubMed.
- C. A. Denard, J. F. Hartwig and H. Zhao, ACS Catalysis, 2013, 3, 2856–2864 CrossRef CAS.
- H. Pellissier, Tetrahedron, 2006, 62, 2143–2173 CrossRef CAS.
- H. E. Schoemaker, D. Mink and M. G. WubboLts, Science, 2003, 299, 1694–1697 CrossRef CAS PubMed.
- K. M. Polizzi, A. S. Bommarius, J. M. Broering and J. F. Chaparro-Riggers, Curr. Opin. Chem. Biol., 2007, 11, 220–225 CrossRef CAS PubMed.
- P. V. Iyer and L. Ananthanarayan, Process Biochem., 2008, 43, 1019–1032 CrossRef CAS.
- M. M. Kristjánsson and J. E. Kinsella, Adv. Food Nutr. Res., 1991, 35, 237–316 Search PubMed.
- C.-L. Tsou, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1995, 1253, 151–162 CrossRef.
- S. Prakash and A. Matouschek, Trends Biochem. Sci., 2004, 29, 593–600 CrossRef CAS PubMed.
- D. J. Brockwell, Curr. Nanosci., 2007, 3, 3–15 CAS.
- T. J. Ahern and A. M. Klibanov, Methods Biochem. Anal., 1988, 33, 91–127 CAS.
- C. Lee and M. Levitt, Nature, 1991, 352, 448–451 CrossRef CAS PubMed.
- K. Yasukawa and K. Inouye, Biochim. Biophys. Acta, Proteins Proteomics, 2007, 1774, 1281–1288 CrossRef CAS PubMed.
- C. P. Govardhan, Curr. Opin. Biotechnol., 1999, 10, 331–335 CrossRef CAS PubMed.
- S. S. Wong and L.-J. C. Wong, Enzyme Microb. Technol., 1992, 14, 866–874 CrossRef CAS PubMed.
- M. L. Villalonga, P. Díez, A. Sánchez, M. Gamella, J. M. Pingarrón and R. Villalonga, Chem. Rev., 2014, 114, 4868–4917 CrossRef CAS PubMed.
- C. Mateo, J. M. Palomo, G. Fernandez-Lorente, J. M. Guisan and R. Fernandez-Lafuente, Enzyme Microb. Technol., 2007, 40, 1451–1463 CrossRef CAS.
- R. A. Sheldon and S. Van Pelt, Chem. Soc. Rev., 2013, 42, 6223–6235 RSC.
- V. G. H. Eijsink, S. GÅseidnes, T. V. Borchert and B. Van Den Burg, Biomol. Eng., 2005, 22, 21–30 CrossRef CAS PubMed.
- A. Gershenson and F. H. Arnold, Genet. Eng, 2000, 22, 55–76 CAS.
- R. Fernandez-Lafuente, Enzyme Microb. Technol., 2009, 45, 405–418 CrossRef CAS.
- D. Brady and J. Jordaan, Biotechnol. Lett., 2009, 31, 1639–1650 CrossRef CAS PubMed; A. S. Bommarius and M. F. Paye, Chem. Soc. Rev., 2013, 42, 6534–6565 RSC.
- S. Cantone, V. Ferrario, L. Corici, C. Ebert, D. Fattor, P. Spizzo and L. Gardossi, Chem. Soc. Rev., 2013, 42, 6262–6276 RSC.
- R. Dicosimo, J. McAuliffe, A. J. Poulose and G. Bohlmann, Chem. Soc. Rev., 2013, 42, 6437–6474 RSC.
- A. Liese and L. Hilterhaus, Chem. Soc. Rev., 2013, 42, 6236–6249 RSC.
- U. Guzik, K. Hupert-Kocurek and D. Wojcieszynska, Molecules, 2014, 19, 8995–9018 CrossRef PubMed.
- E. T. Hwang and M. B. Gu, Eng. Life Sci., 2013, 13, 49–61 CrossRef CAS.
- C. Garcia-Galan, A. Berenguer-Murcia, R. Fernandez-Lafuente and R. C. Rodrigues, Adv. Synth. Catal., 2011, 353, 2885–2904 CrossRef CAS.
- O. Barbosa, C. Ortiz, Á. Berenguer-Murcia, R. Torres, R. C. Rodrigues and R. Fernandez-Lafuente, Biotechnol. Adv., 2015, 33, 435–456 CrossRef CAS PubMed.
- J. Köditz, R. Ulbrich-Hofmann and U. Arnold, Eur. J. Biochem., 2004, 271, 4147–4156 CrossRef PubMed.
- U. Arnold and R. Ulbrich-Hofmann, J. Protein Chem., 2000, 19, 345–352 CrossRef CAS PubMed.
- R. Ulbrich-Hofmann, U. Arnold and J. Mansfeld, J. Mol. Catal. B: Enzym., 1999, 7, 125–131 CrossRef CAS.
- J. Mansfeld, G. Vriend, B. Van Den Burg, V. G. H. Eijsink and R. Ulbrich-Hofmann, Biochemistry, 1999, 38, 8240–8245 CrossRef CAS PubMed.
- V. Grazu, F. López-Gallego, T. Montes, O. Abian, R. González, J. A. Hermoso, J. L. García, C. Mateo and J. M. Guisán, Process Biochem., 2010, 45, 390–398 CrossRef CAS.
- C. A. Godoy, B. D. L. Rivas, V. Grazú, T. Montes, J. M. Guisàn and F. López-Gallego, Biomacromolecules, 2011, 12, 1800–1809 CrossRef CAS PubMed.
- F. Secundo, Chem. Soc. Rev., 2013, 42, 6250–6261 RSC.
- H. Yang, S. Yang, J. Kong, A. Dong and S. Yu, Nat. Protoc., 2015, 10, 382–396 CrossRef CAS PubMed.
- H. Zhao, C. L. Jones and J. V. Cowins, Green Chem., 2009, 11, 1128–1138 RSC.
- D. M. Byler, J. N. Brouillette and H. Susi, Spectroscopy, 1986, 1, 29–32 CAS.
- R. M. Lau, M. J. Sorgedrager, G. Carrea, F. Van Rantwijk, F. Secundo and R. A. Sheldon, Green Chem., 2004, 6, 483–487 RSC.
- K. J. Halverson, I. Sucholeiki, T. T. Ashburn and P. T. Lansbury Jr, J. Am. Chem. Soc., 1991, 113, 6701–6703 CrossRef CAS.
- D. F. Izquierdo, O. Barbosa, M. Isabel Burguete, P. Lozano, S. V. Luis, R. Fernandez-Lafuente and E. García-Verdugo, RSC Adv., 2014, 4, 6219–6225 RSC.
- J. C. S. D. Santos, O. Barbosa, C. Ortiz, A. Berenguer-Murcia, R. C. Rodrigues and R. Fernandez-Lafuente, ChemCatChem, 2015, 7, 2413–2432 CrossRef.
- C. Mateo, J. M. Palomo, M. Fuentes, L. Betancor, V. Grazu, F. López-Gallego, B. C. C. Pessela, A. Hidalgo, G. Fernández-Lorente, R. Fernández-Lafuente and J. M. Guisán, Enzyme Microb. Technol., 2006, 39, 274–280 CrossRef CAS.
- C. Mateo, O. Abian, M. Bernedo, E. Cuenca, M. Fuentes, G. Fernandez-Lorente, J. M. Palomo, V. Grazu, B. C. C. Pessela, C. Giacomini, G. Irazoqui, A. Villarino, K. Ovsejevi, F. Batista-Viera, R. Fernandez-Lafuente and J. M. Guisán, Enzyme Microb. Technol., 2005, 37, 456–462 CrossRef CAS.
- J. Pedroche, M. del Mar Yust, C. Mateo, R. Fernández-Lafuente, J. Girón-Calle, M. Alaiz, J. Vioque, J. M. Guisán and F. Millán, Enzyme Microb. Technol., 2007, 40, 1160–1166 CrossRef CAS.
- C. M. A. Galvao, A. F. Souza Silva, M. F. Custódio, R. Monti and R. L. C. Giordano, Controlled hydrolysis of cheese whey proteins using trypsin and α-chymotrypsin, Appl. Biochem. Biotechnol., Part A, 2001, 91–93, 761–776 CrossRef CAS.
- J. Pedroche, M. M. Yust, H. Lqari, C. Megias, J. Girón-Calle, M. Alaiz, J. Vioque and F. Millán, Food Res. Int., 2007, 40, 931–938 CrossRef CAS.
- J. Pedroche, M. M. Yust, H. Lqari, J. Girón-Calle, J. Vioque, M. Alaiz and F. Millán, Int. Dairy J., 2004, 14, 527–533 CrossRef CAS.
- J. Delfín, I. Martínez, W. Antuch, V. Morera, Y. González, R. Rodríguez, M. Márquez, A. Saroyán, N. Larionova, J. Díaz, G. Padrón and M. Chávez, Toxicon, 1996, 34, 1367–1376 CrossRef.
- J. Delfin, Y. Gonzalez, J. Diaz and M. Chavez, Arch. Med. Res., 1994, 25, 199–204 CAS.
- J. C. S. Dos Santos, N. Rueda, O. Barbosa, M. D. C. Millán-Linares, J. Pedroche, M. Del Mar Yuste, L. R. B. Gonçalves and R. Fernandez-Lafuente, J. Mol. Catal. B: Enzym., 2015, 117, 38–44 CrossRef CAS.
- R. M. Blanco and J. M. Guisán, Enzyme Microb. Technol., 1988, 10, 227–232 CrossRef CAS.
- (a) J. Ge, J. Lei and R. N. Zare, Nat. Nanotechnol., 2012, 7, 428–432 CrossRef CAS PubMed; (b) F. Lyu, Y. Zhang, R. N. Zare, J. Ge and Z. Liu, Nano Lett., 2014, 14, 5761–5765 CrossRef CAS PubMed; (c) Z. Li, Y. Zhang, Y. Su, P. Ouyang, J. Ge and Z. Liu, Chem. Commun., 2014, 50, 12465–12468 RSC; (d) X. Wu, M. Hou and J. Ge, Catal. Sci. Technol., 2015, 5, 5077–5085 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra03627a |
| ‡ Both authors have evenly contributed to this paper. |
| This journal is © The Royal Society of Chemistry 2016 |