
Construction and luminescence property of a highly ordered 2D self-assembled amphiphilic bidentate organoplatinum(ii) complex†
Abstract
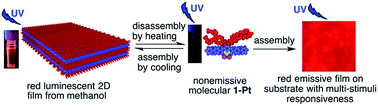
Rod-coil shaped amphiphilic bidentate dichloro(phenanthroline)platinum(II) complexes, 1-Pt and 2-Pt, which possess different hydrophilic lateral chains on one side of an identical aromatic rod core, are synthesized. 1-Pt first forms dimers then arranges into well-defined two-dimensional (2D) films consisting of highly ordered molecular arrays both from methanol and on substrate. 2-Pt, as a building-block, yields micrometer-sized rigid 2D sheets without formation of an initial dimer. Red luminescence of the film is induced by self-assembly of nonemissive 1-Pt molecules, whereas sheets based on nonemissive 2-Pt gives weak yellow emission. These results indicate that the coil-rod ratio plays an important role in the structure and optical properties of these self-assemblies. Moreover, the film on the substrate at the macroscopic scale, exhibits multi-stimuli responsiveness, which predicts its application in smart chemosensing devices and probes.
- This article is part of the themed collection: Luminescence and photophysical properties of metal complexes
 Please wait while we load your content...
Please wait while we load your content...

