
Tacrine-allyl/propargylcysteine–benzothiazole trihybrids as potential anti-Alzheimer's drug candidates†
Abstract
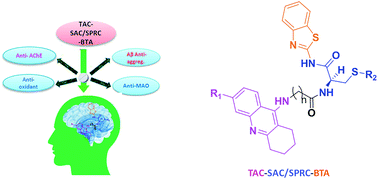
On continuing our research into new drug candidates for Alzheimer's disease (AD), we have designed, synthesized and evaluated a series of multifunctional trihybrid agents. The design strategy was based on the incorporation of a benzothiazole (BTA) moiety on a series of very recently reported bihybrids, resulting from the conjugation of a tacrine (TAC) with natural based moieties, namely S-allylcysteine (SAC) (garlic constituent) and S-propargylcysteine (SPRC). Thus, in addition to the acetylcholinesterase inhibition (AChEI) and anti-ROS capacity of the bihybrids (TAC–SAC/SPRC), the new trihybrids (TAC–SAC/SPRC–BTA) appeared endowed with a 5-fold capacity for inhibition of amyloid beta-peptide (Aβ) aggregation. The BTA moiety led also to considerable enhancement of the AChEI on the trihybrids, which molecular modeling suggested as being due to simultaneous binding to the catalytic active site and peripheral anionic site of AChE. The trihybrids were also assessed for MAO inhibition, but resulted in lower activity than expected, ascribed to the low accessibility of the propargyl groups to the enzyme active site. Finally, the effects of the compounds on the viability of neuroblastome cells stressed with Aβ42 and H2O2 showed moderate cell protection. Overall, the performed studies illustrate the importance (and limitations) of enclosing several molecular scaffolds in one molecular entity to allow the modulation of multiple AD targets.
- This article is part of the themed collection: Towards understanding and treating Alzheimer’s disease
 Please wait while we load your content...
Please wait while we load your content...

