
Exceptional activity of gallium(iii) chloride and chlorogallate(iii) ionic liquids for Baeyer–Villiger oxidation†
Abstract
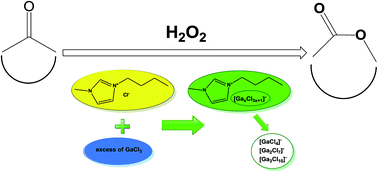
Baeyer–Villiger oxidation of cyclic ketones, using H2O2 as the oxidising agent, was systematically studied using a range of metal chlorides in different solvents, and in neat chlorogallate(III) ionic liquids. The extremely high activity of GaCl3 in promoting oxidation with H2O2, irrespective of solvent, was reported for the first time. The activity of all other metal chlorides was strongly solvent-dependent. In particular, AlCl3 was very active in a protic solvent (ethanol), and tin chlorides, SnCl4 and SnCl2, were active in aprotic solvents (toluene and dioxane). In order to eliminate the need for volatile organic solvent, a Lewis acidic chlorogallate(III) ionic liquid was used in the place of GaCl3, which afforded typically 89–94% yields of lactones in 1–120 min, at ambient conditions. Raman and 71Ga NMR spectroscopic studies suggest that the active species, in both GaCl3 and chlorogallate(III) ionic liquid systems, are chlorohydroxygallate(III) anions, [GaCl3OH]−, which are the products of partial hydrolysis of GaCl3 and chlorogallate(III) anions; therefore, the presence of water is crucial.
 Please wait while we load your content...
Please wait while we load your content...

