
Transition-metal free alkylarylation of acrylamides initiated by radical C–C bond cleavage of the tertiary cycloalkanols†
Li-Na Guo*a,
Zhi-Qiang Denga,
Yong Wua and
Jie Hub
aDepartment of Chemistry, School of Science and MOE Key Laboratory for Nonequilibrium Synthesis and Modulation of Condensed Matter, Xi'an Jiaotong University, Xi'an 710049, China. E-mail: guoln81@xjtu.edu.cn
bXi'an Libang Pharmaceutical Co., LTD, China
First published on 7th March 2016
Abstract
An efficient Na2S2O8-promoted radical cyclization reaction of acrylamides with the tertiary cycloalkanols has been developed. This one pot procedure involves a tandem C–C bond cleavage and two C–C bonds formation process. The key advantages of this protocol are its transition-metal free, its operational simplicity and its excellent functional group tolerance, thus allowing a facile access to oxindoles containing a carbonyl group.
Over the past few years, transition-metal-free coupling reactions have been developed as attractive tools for carbon–carbon and carbon–heteroatom bonds formation and become an enabling strategy for sustainable synthesis.1 In this field, transition-metal-free oxidative coupling reactions have attracted significant attention and different types of oxidants including hypervalent iodine reagents, DDQ, TEMPO and others have been utilized successfully to promote the oxidative coupling.1 In recent years, extensive efforts have been made towards the use of persulfates as efficient oxidants in the oxidative coupling reactions due to their low toxicity, easily handling and commercial availability.2 In this aspect, our group has demonstrated K2S2O8 promoted spirocyclization of hydroxymethylacrylamide with 1,3-dicarbonyl compounds, which provides a mild and efficient access to novel and unavailable spirooxindoles.3a Recently, we have also presented the oxidative tandem radical thiocyanooxygenation of olefinic amides for the synthesis of SCN-containing benzoxazines and imino-isobenzofurans using K2S2O8 as the sole oxidant.3b Although, some progress have been made, it is still highly desirable to explore the application of persulfates for oxidative coupling reactions, especially for the synthesis of other structurally diverse heterocycles.1
Oxindoles are a class of important structural motifs found in a wide range of bioactive natural products and pharmaceutical molecules. Moreover, they also serve as important building blocks in synthetic chemistry and drug design.4 Thus, the development of new and simple methods for constructing this privileged scaffold is highly desirable and has attracted considerable attentions.5 Recently, the radical-mediated cyclization of N-arylacrylamides with different reactants has emerged as an efficient and straightforward approach to obtain various functionalized oxindoles.6 All of these presented methods are based on the addition of radicals to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond of N-arylacrylamides followed by an intramolecular radical substitution to achieve this skeleton. Recently, our group has demonstrated that the strained tertiary cycloalkanols could easily undergo a radical ring-opening coupling with alkynyl hypervalent iodide in the presence of persulfate, in which an alkyl radical intermediate bearing a carbonyl group was involved.3e,7 Inspired by this result and combined with our ongoing research interests in the radical-mediated cyclization,8 we envisioned that the strained tertiary cycloalkanols might be a suitable substrate to initiate the radical cyclization of N-arylacrylamides under mild conditions. Herein, we wish to report a transition-metal free tandem radical cyclization of N-arylacrylamides with the strained tertiary cycloalkanols.9 In this tandem reaction, the cyclization process is triggered by a free radical generated by an oxidative ring-opening process.
C bond of N-arylacrylamides followed by an intramolecular radical substitution to achieve this skeleton. Recently, our group has demonstrated that the strained tertiary cycloalkanols could easily undergo a radical ring-opening coupling with alkynyl hypervalent iodide in the presence of persulfate, in which an alkyl radical intermediate bearing a carbonyl group was involved.3e,7 Inspired by this result and combined with our ongoing research interests in the radical-mediated cyclization,8 we envisioned that the strained tertiary cycloalkanols might be a suitable substrate to initiate the radical cyclization of N-arylacrylamides under mild conditions. Herein, we wish to report a transition-metal free tandem radical cyclization of N-arylacrylamides with the strained tertiary cycloalkanols.9 In this tandem reaction, the cyclization process is triggered by a free radical generated by an oxidative ring-opening process.
Initially, N-phenyl acrylamide 1a and the tertiary cyclopropanol 2a were used as the model substrates to screen optimal conditions (Table 1). To our delight, the desired γ-carbonyl alkyl substituted oxindole 3a was isolated in 46% yield in the presence of 1.5 equiv. of K2S2O8 in 2 mL of acetone/H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) (entry 1). Other homogeneous systems were also effective for this reaction and the co-solvent consisting of AcOH/H2O (1:1) gave the best result, affording the 3a in 76% yield (entries 2–5). However, when heterogeneous solvents such as CH2Cl2/H2O and PhCl/H2O were used, no desired product 3a was observed (entries 6 and 7). Notably, H2O was also suitable solvent for this reaction, albeit in 26% yield (entry 8). Applying AcOH as the sole solvent resulted in a trace amount of 3a, which implied that water played an important role in this transformation (entry 9). Adjusting the ratio of AcOH and H2O did not improve the yield (entries 10 and 11). Among different oxidants investigated, Na2S2O8 proved to be the best oxidant (entries 12–14). Unfortunately, raising or reducing the amount of Na2S2O8 could not enhance the yield further (entries 15 and 16). Finally, no reaction was observed in the absence of oxidant (entry 17).
:1) (entry 1). Other homogeneous systems were also effective for this reaction and the co-solvent consisting of AcOH/H2O (1:1) gave the best result, affording the 3a in 76% yield (entries 2–5). However, when heterogeneous solvents such as CH2Cl2/H2O and PhCl/H2O were used, no desired product 3a was observed (entries 6 and 7). Notably, H2O was also suitable solvent for this reaction, albeit in 26% yield (entry 8). Applying AcOH as the sole solvent resulted in a trace amount of 3a, which implied that water played an important role in this transformation (entry 9). Adjusting the ratio of AcOH and H2O did not improve the yield (entries 10 and 11). Among different oxidants investigated, Na2S2O8 proved to be the best oxidant (entries 12–14). Unfortunately, raising or reducing the amount of Na2S2O8 could not enhance the yield further (entries 15 and 16). Finally, no reaction was observed in the absence of oxidant (entry 17).
|
|||
|---|---|---|---|
| Entry | Oxidant (equiv.) | Solvent | Yieldb (%) |
| a Reaction conditions: 1a (0.2 mmol, 1 equiv.), 2a (0.3 mmol, 1.5 equiv.), solvent (2 mL), oxidant (1.5 equiv.), 50 °C, 24 h, under N2.b Yield of isolated product.c n.r. = no reaction.d Yield on a 1 mmol scale is given in parentheses. | |||
| 1 | K2S2O8 (1.5) | Acetone/H2O (1:1) |
46 |
| 2 | K2S2O8 (1.5) | CH3CN/H2O (1:1) |
43 |
| 3 | K2S2O8 (1.5) | DMF/H2O (1:1) |
30 |
| 4 | K2S2O8 (1.5) | DMSO/H2O (1:1) |
29 |
| 5 | K2S2O8 (1.5) | AcOH/H2O (1:1) |
76 |
| 6 | K2S2O8 (1.5) | CH2Cl2/H2O (1:1) |
n.r.c |
| 7 | K2S2O8 (1.5) | PhCl/H2O (1:1) |
n.r.c |
| 8 | K2S2O8 (1.5) | H2O | 26 |
| 9 | K2S2O8 (1.5) | AcOH | Trace |
| 10 | K2S2O8 (1.5) | AcOH/H2O (1:3) |
68 |
| 11 | K2S2O8 (1.5) | AcOH/H2O (3:1) |
65 |
| 12 | Na2S2O8 (1.5) | AcOH/H2O (1:1) |
82 (80)d |
| 13 | (NH4)2S2O8 (1.5) | AcOH/H2O (1:1) |
50 |
| 14 | Oxone (1.5) | AcOH/H2O (1:1) |
16 |
| 15 | Na2S2O8 (1.0) | AcOH/H2O (1:1) |
31 |
| 16 | Na2S2O8 (2.0) | AcOH/H2O (1:1) |
42 |
| 17 | — | AcOH/H2O (1:1) |
n.r.c |
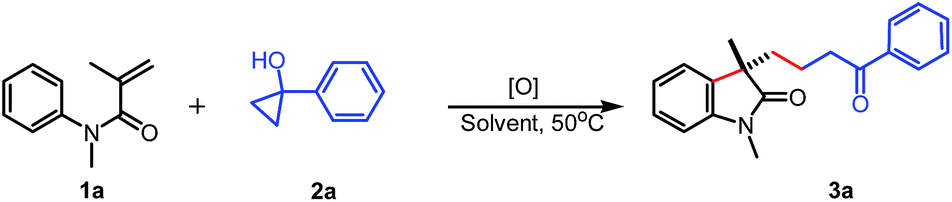
With the optimized reaction conditions in hand, the scope of cyclopropanols was surveyed with acrylamide 1a. As shown in Table 2, a variety of cyclopropanols containing electron-withdrawing or -donating group at the para position of aromatic ring were all successfully engaged in this reaction (3b–d). Cyclopropanols with a strong electron-withdrawing trifluoromethyl group could also be survived in the reaction, giving the corresponding product 3e in 54% yield. In addition, it was found that the 1-benzyl cyclopropanol was also good substrate, affording the corresponding product 3f in 80% yield. The benzylic cyclopropanols containing a But, OMe, or Br, at para position of the aromatic ring also worked well to give the corresponding oxindoles 3g–i in good yields. Finally, this reaction was not limited to aromatic and benzylic substituted cyclopropanols. Cyclopropanols with different alkyl groups were also suitable substrates. For example, the cyclopropanols 2j and 2k underwent the cyclization process to produce 3j and 3k in 73% and 40% yields, respectively. Satisfactorily, the reaction of 1-phenoxymethylcyclopropanol 2l with 1a also led to the desired oxindole 3l in 80% yield.
|
|---|
 |
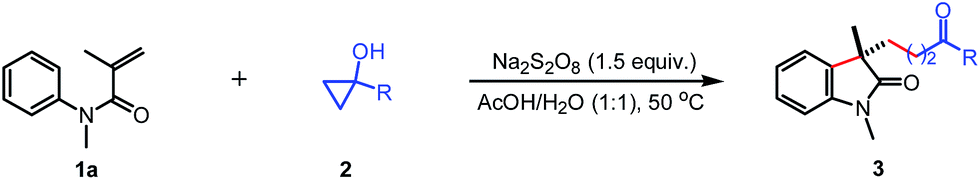
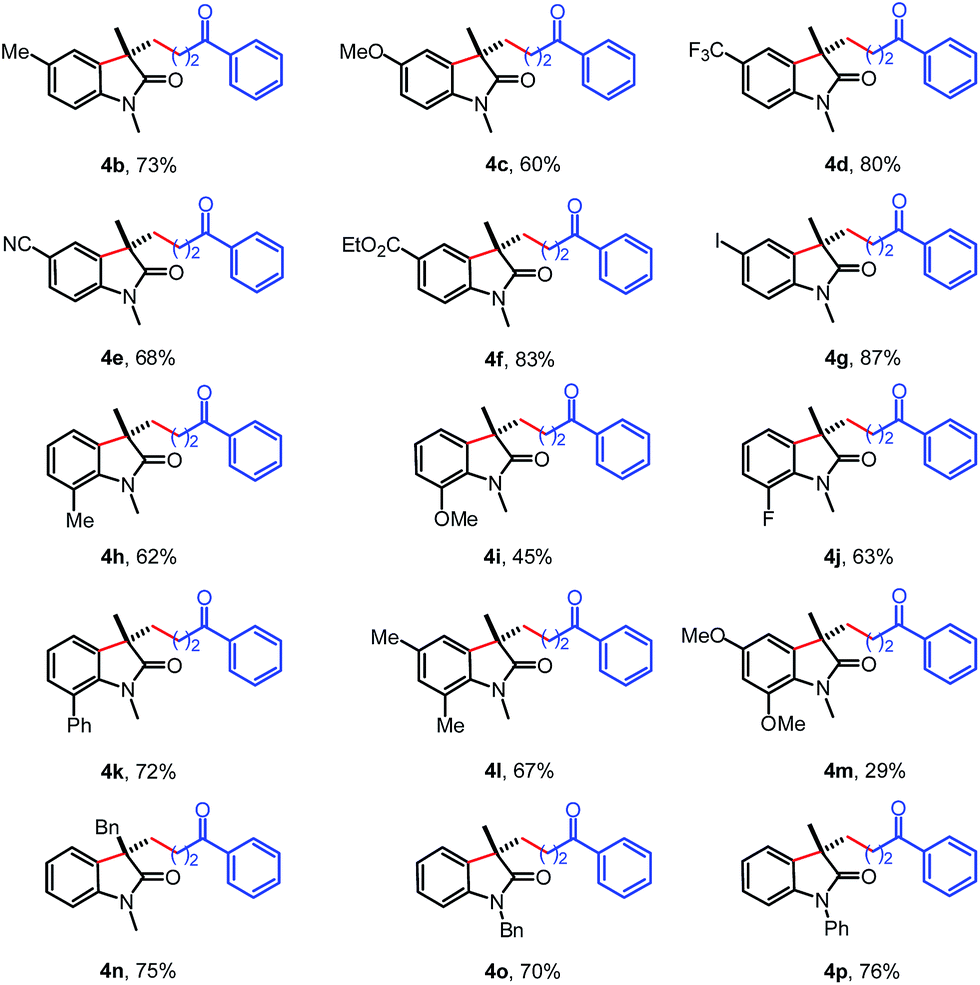
The scope of the N-arylacrylamides 1 was examined next (Table 3). It was found that the electron-donating groups (Me, MeO) and electron-withdrawing groups (CF3, CN, CO2Et, I) on the aniline moiety were tolerated under these mild reaction conditions (4b–4g). The sterically congested o-substituted acrylamides also worked with tertiary cyclopropanol 2a to deliver the corresponding products in moderate yields (4h–k). The multisubstituted substrate bearing two methyl groups gave the desired oxindole 4l in 67% yield. While only 29% yield was isolated for substrate containing two methoxyl groups due to the low conversion. Satisfactorily, the analogous acrylamide 1l with a benzyl group at the α-position also furnished the desired product 4n in 75% yield. Besides to the methyl group, the acrylamide bearing a benzyl or phenyl protecting group on the N tether also afforded the oxindole 4o and 4p in 70% and 76% yields, respectively.
|
|---|
 |
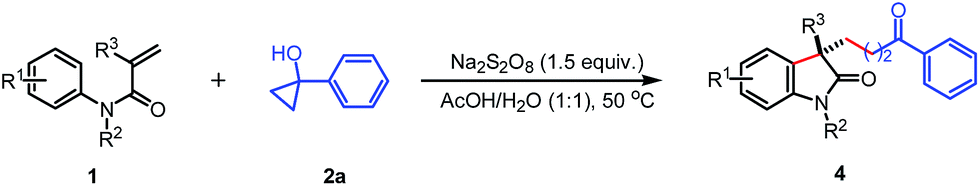
Finally, the tertiary cyclobutanols were subjected to this reaction, and the corresponding alkyl substituted oxindoles 6a–c were obtained in 40–44% yields (eqn (1)), respectively. The low yield is attributed to the formation of the byproduct 1-tetralone which was formed by the radical ring-expansion of cyclobutanols.7k Unfortunately, the less strained tertiary cyclopentanols failed to give the desired products.
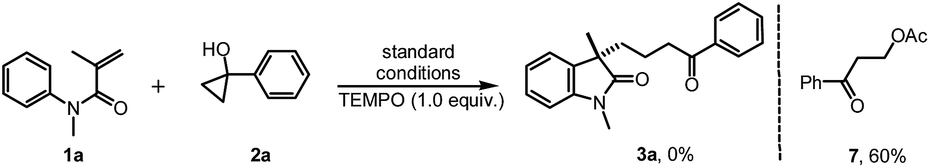
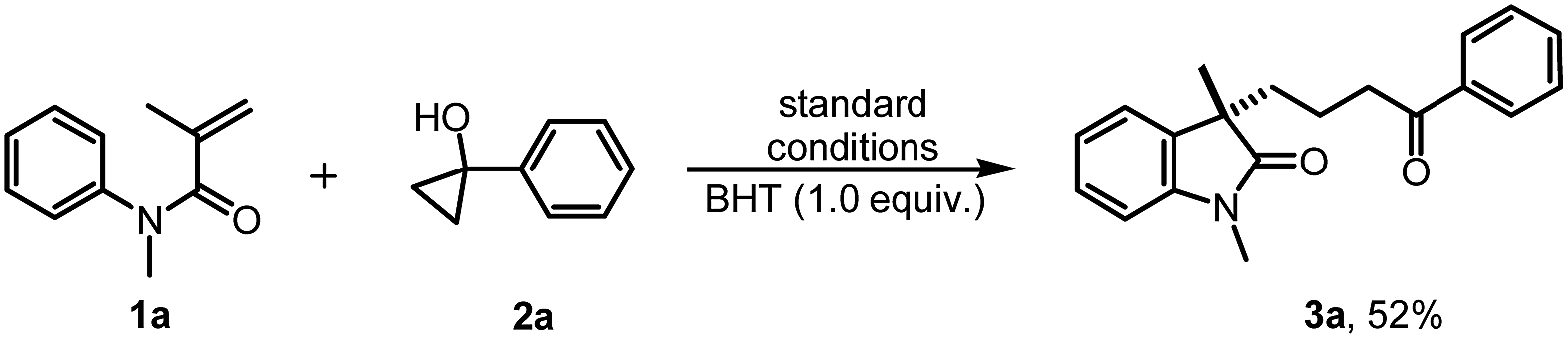
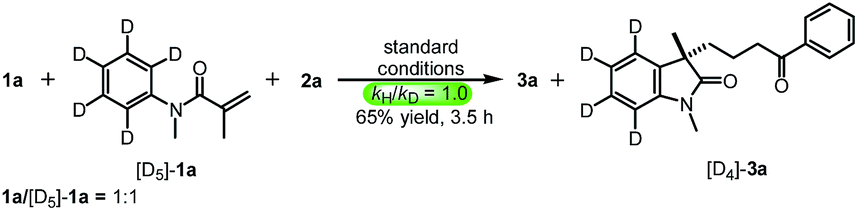
To gain insight into the mechanism of this reaction, several mechanistic experiments were carried out (for details, see the ESI†). Firstly, we found that the reaction of 1a with 2a was suppressed remarkably by addition of radical scavenger TEMPO or BHT, respectively (eqn (2) and (3)). When a stoichiometric amount of TEMPO was added, no desired product 3a was observed along with a coupling product 7 isolated. The result implies that the reaction may proceed via a radical pathway. Next, the intra- and intermolecular kinetic isotope effect (KIE) experiments were also performed, respectively. A negligible KIE (kH/kD = 1.0) for the intra- and intermolecular competition experiments were observed, respectively (eqn (4) and (5)). On the basis of these mechanistic studies and previous reports,6,7 we propose a mechanism for this tandem cyclization reaction as outlined in Scheme 1.10 Firstly, the β-keto radical II was formed by C–C bond cleavage of cyclopropanol 2a and rearrangement in the presence of oxidant.11 Secondly, the radical II subsequent adds to the CC bond of acrylamide 1a to generate the intermediate III, followed by intramolecular radical substitution to give the product 3a.
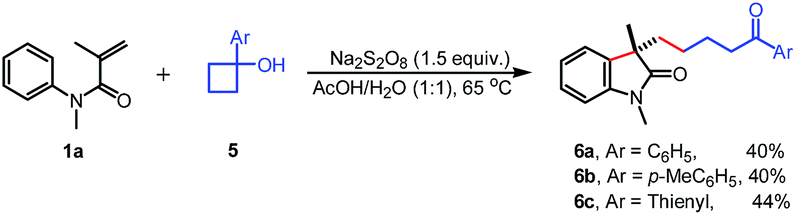 | (1) |
 | (2) |
 | (3) |
 | (4) |
 | (5) |
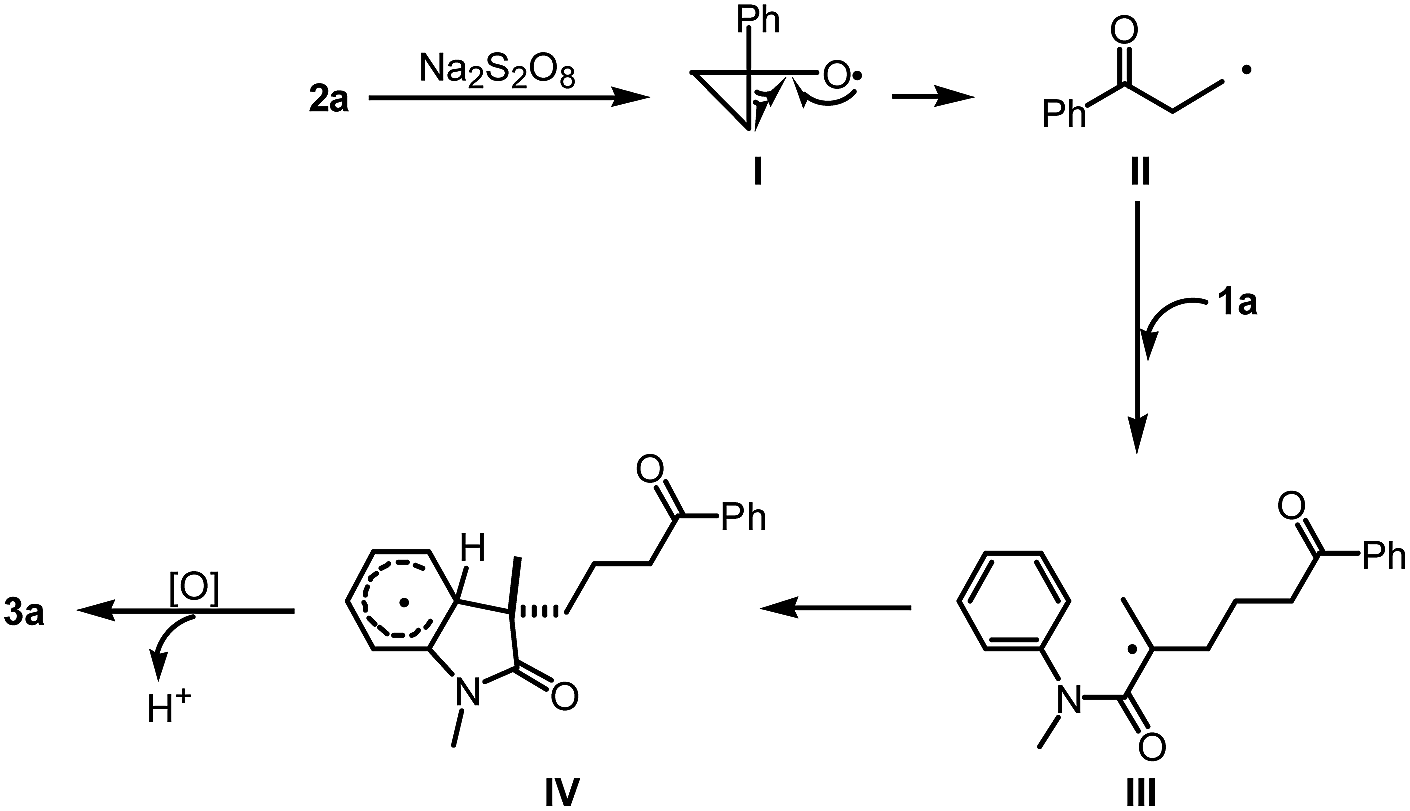 | ||
| Scheme 1 Proposed mechanism. | ||
In summary, we have demonstrated a transition-metal free tandem radical cyclization of acrylamides with tertiary cycloalkanols under very mild conditions. In this tandem transformation, the oxidative ring-opening of cycloalkanols initiated a difunctionalization of activated alkenes to achieved cyclization. This strategy allows a facile accessing to a serious of oxindoles containing a remote carbonyl group.
Acknowledgements
Financial support from Natural Science Basic Research Plan in Shaanxi Province of China (No. 2014JQ2071) and the Fundamental Research Funds of the Central Universities (No. 2015qngz17) are greatly appreciated.Notes and references
- For reviews, see: (a) J. J. Mousseau and A. B. Charette, Acc. Chem. Res., 2013, 46, 412 CrossRef CAS PubMed; (b) C.-L. Sun and Z.-J. Shi, Chem. Rev., 2014, 114, 9219 CrossRef CAS PubMed.
- (a) X. Li, H.-Y. Wang and Z.-J. Shi, New J. Chem., 2013, 37, 1704 RSC; (b) Y.-M. Li, X.-H. Wei, X.-A. Li and S.-D. Yang, Chem. Commun., 2013, 49, 11701 RSC; (c) Y.-M. Li, Y. Shen, K.-J. Chang and S.-D. Yang, Tetrahedron, 2014, 70, 1991 CrossRef CAS; (d) W. Wei, J. Wen, D. Yang, J. Du, J. You and H. Wang, Green Chem., 2014, 16, 2988 RSC; (e) W. Wei, J. Wen, D. Yang, X. Liu, M. Guo, R. Dong and H. Wang, J. Org. Chem., 2014, 79, 4225 CrossRef CAS PubMed; (f) J. K. Laha, K. S. S. Tummalapalli and A. Gupta, Org. Lett., 2014, 16, 4392 CrossRef CAS PubMed; (g) F. Yin and X.-S. Wang, Org. Lett., 2014, 16, 1128 CrossRef CAS PubMed.
- (a) H. Wang, L.-N. Guo and X.-H. Duan, Org. Lett., 2013, 15, 5254 CrossRef CAS PubMed; (b) H. Yang, X.-H. Duan, J.-F. Zhao and L.-N. Guo, Org. Lett., 2015, 17, 1998 CrossRef CAS PubMed. For other examples concerning transition-metal-free coupling and cyclization reactions have developed in our group, see: (c) S. Wang, L.-Y. He and L.-N. Guo, Synthesis, 2015, 3191 CAS; (d) H. Wang, L.-N. Guo, S. Wang and X.-H. Duan, Org. Lett., 2015, 17, 3054 CrossRef CAS PubMed; (e) S. Wang, L.-N. Guo, H. Wang and X.-H. Duan, Org. Lett., 2015, 17, 4798 CrossRef CAS PubMed; (f) J.-F. Zhao, X.-H. Duan, H. Yang and L.-N. Guo, J. Org. Chem., 2015, 80, 11149 CrossRef CAS PubMed; (g) H. Wang, H. Yang, Y. Li and X.-H. Duan, RSC Adv., 2014, 4, 8720 RSC; (h) S. Zhang, L.-N. Guo, H. Wang and X.-H. Duan, Org. Biomol. Chem., 2013, 11, 4308 RSC.
- For reviews, see: (a) C. Marti and E. M. Carreira, Eur. J. Org. Chem., 2003, 2209 CrossRef CAS; (b) H. Lin and S. J. Danishefsky, Angew. Chem., Int. Ed., 2003, 42, 36 CrossRef CAS; (c) C. V. Galliford and K. A. Scheidt, Angew. Chem., Int. Ed., 2007, 46, 8748 CrossRef CAS PubMed; (d) G. S. Singh and Z. Y. Desta, Chem. Rev., 2012, 112, 6104 CrossRef CAS PubMed; (e) K. Shen, X. Liu, L. Lin and X. Feng, Chem. Sci., 2012, 3, 327 RSC; (f) R. Dalpozzo, G. Bartoli and G. Bencivenni, Chem. Soc. Rev., 2012, 41, 7247 RSC; (g) N. R. Ball-Jones, J. J. Badillo and A. K. Franz, Org. Biomol. Chem., 2012, 10, 5165 RSC; (h) L. Hong and R. Wang, Adv. Synth. Catal., 2013, 355, 1023 CrossRef CAS.
- For recent reviews, see: (a) F. Zhou, Y.-L. Liu and J. Zhou, Adv. Synth. Catal., 2010, 352, 1381 CrossRef CAS; (b) J. E. M. N. Klein and R. J. K. Taylor, Eur. J. Org. Chem., 2011, 6821 CrossRef CAS.
- For reviews, see: (a) J.-R. Chen, X.-Y. Yu and W.-J. Xiao, Synthesis, 2015, 604 Search PubMed; (b) R.-J. Song, Y. Liu, Y.-X. Xie and J.-H. Li, Synthesis, 2015, 1195 CAS; for selected examples, see: (c) T. Wu, H. Zhang and G. Liu, Tetrahedron, 2012, 68, 5229 CrossRef CAS; (d) W.-T. Wei, M.-B. Zhou, J.-H. Fan, W. Liu, R.-J. Song, Y. Liu, M. Hu, P. Xie and J.-H. Li, Angew. Chem., Int. Ed., 2013, 52, 3638 CrossRef CAS PubMed; (e) Y.-M. Li, M. Sun, H.-L. Wang, Q.-P. Tian and S.-D. Yang, Angew. Chem., Int. Ed., 2013, 52, 3972 CrossRef CAS PubMed; (f) J. Xie, P. Xu, H. Li, Q. Xue, H. Jin, Y. Cheng and C. Zhu, Chem. Commun., 2013, 49, 5672 RSC; (g) X. Li, X. Xu, P. Hu, X. Xiao and C. Zhou, J. Org. Chem., 2013, 78, 7343 CrossRef CAS PubMed; (h) K. Matcha, R. Narayan and A. P. Antonchick, Angew. Chem., Int. Ed., 2013, 52, 7985 CrossRef CAS PubMed; (i) T. Shen, Y. Yuan and N. Jiao, Chem. Commun., 2014, 50, 554 RSC; (j) Z. Li, Y. Zhang, L. Zhang and Z.-Q. Liu, Org. Lett., 2014, 16, 382 CrossRef CAS PubMed.
- For selected examples on tandem radical ring-opening coupling of cycloalkanols involving transition-metal catalysts, see: (a) K. Meyer and J. Roček, J. Am. Chem. Soc., 1972, 94, 1209 CrossRef CAS; (b) N. Iwasawa, S. Hayakawa, M. Funahashi, K. Isobe and K. Narasaka, Bull. Chem. Soc. Jpn., 1993, 66, 819 CrossRef CAS; (c) N. Iwasawa, M. Funahashi, S. Hayakawa, T. Ikeno and K. Narasaka, Bull. Chem. Soc. Jpn., 1999, 72, 85 CrossRef CAS; (d) S. Chiba, Z. Cao, S. A. A. E. Bialy and K. Narasaka, Chem. Lett., 2006, 18 CrossRef CAS; (e) J. Jiao, L. X. Nguyen, D. R. Patterson and R. A. Flowers II, Org. Lett., 2007, 9, 1323 CrossRef CAS PubMed; (f) A. Ilangovan, S. Saravanakumar and S. Malayappasamy, Org. Lett., 2013, 15, 4968 CrossRef CAS PubMed; (g) Y.-F. Wang and S. Chiba, J. Am. Chem. Soc., 2009, 131, 12570 CrossRef CAS PubMed; (h) H. Zhao, X. Fan, J. Yu and C. Zhu, J. Am. Chem. Soc., 2015, 137, 3490 CrossRef CAS PubMed; (i) S. Ren, C. Feng and T.-P. Loh, Org. Biomol. Chem., 2015, 13, 5105 RSC; (j) R. Ren, H. Zhao, L. Huan and C. Zhu, Angew. Chem., Int. Ed., 2015, 54, 12692 CrossRef CAS PubMed; (k) J. Yu, H. Zhao, S. Liang, X. Bao and C. Zhu, Org. Biomol. Chem., 2015, 13, 7924 RSC; for photocatalyzed ring opening of cyclopropanols, see: (l) S. Bloom, D. D. Bume, C. R. Pitts and T. Lectka, Chem.–Eur. J., 2015, 21, 8060 CrossRef CAS PubMed.
- (a) H. Wang, L.-N. Guo and X.-H. Duan, Adv. Synth. Catal., 2013, 355, 2222 CrossRef CAS; (b) Y. Meng, L.-N. Guo, H. Wang and X.-H. Duan, Chem. Commun., 2013, 49, 7540 RSC; (c) S.-L. Zhou, L.-N. Guo, H. Wang and X.-H. Duan, Chem.–Eur. J., 2013, 19, 12970 CrossRef CAS PubMed; (d) H. Wang, L.-N. Guo and X.-H. Duan, Chem. Commun., 2013, 49, 10370 RSC; (e) S.-L. Zhou, L.-N. Guo, S. Wang and X.-H. Duan, Chem. Commun., 2014, 50, 3589 RSC; (f) H. Yang, L.-N. Guo and X.-H. Duan, RSC Adv., 2014, 4, 52986 RSC; (g) L.-N. Guo, S. Wang, X.-H. Duan and S.-L. Zhou, Chem. Commun., 2015, 51, 4803 RSC.
- During the completion of our work on this chemistry, a similar work on Ag-catalyzed oxidative/cyclization of acrylamides with cyclopropanols was reported by Yang, et al. For more details, see: Y. Li, J. Wang, X. Wei and S. Yang, Chin. J.org. Chem., 2015, 35, 638 CrossRef CAS.
- For related examples concerning Ti(III) catalyzed radical arylation of epoxide, see: (a) A. Gansäuer, M. Behlendorf, D. von Laufenberg, A. Fleckhaus, C. Kube, D. V. Sadasivam and R. A. Flowers II, Angew. Chem., Int. Ed., 2012, 51, 4739 CrossRef PubMed; (b) A. Gansäuer, S. Hildebrandt, A. Michelmann, T. Dahmen, D. von Laufenberg, C. Kube, G. D. Fianu and R. A. Flowers II, Angew. Chem., Int. Ed., 2015, 54, 7003 CrossRef PubMed; (c) A. Gansäuer, D. von Laufenberg, C. Kube, T. Dahmen, A. Michelmann, M. Behlendorf, R. Sure, M. Seddiqzai, S. Grimme, D. V. Sadasivam, G. D. Fianu and R. A. Flowers II, Chem.–Eur. J., 2015, 21, 280 CrossRef PubMed.
- For the example on transition-metal free radical trifluoromethylation of heterocycles, see: Y. Ji, T. Brueckl, R. D. Baxter, Y. Fujiwara, I. B. Seiple, S. Su, D. G. Blackmond and P. S. Baran, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 14411 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra03431d |
| This journal is © The Royal Society of Chemistry 2016 |