DOI:
10.1039/C6RA03420A
(Paper)
RSC Adv., 2016,
6, 43697-43706
Silver nanoparticles modified reduced graphene oxide wrapped Ag3PO4/TiO2 visible-light-active photocatalysts with superior performance†
Received
5th February 2016
, Accepted 28th April 2016
First published on 29th April 2016
Abstract
Silver nanoparticles (Ag NPs) modified reduced graphene oxide wrapped Ag3PO4/TiO2 (Ag3PO4/TiO2/Ag-rGO, Ag-ATG) photocatalysts have been developed through a rational design that combines the optimization of charge generation, separation and transfer in the composites, which helps to increase the photocatalytic performance. The Ag-ATG composites possess novel microstructure, in which TiO2 mesoporous spheres of hundreds of nanometers in size are decorated with dense nano-sized Ag3PO4 to form pinecone-liked Ag3PO4/TiO2 particles, which were further wrapped by rGO sheets that are selectively decorated with Ag NPs. The Ag-ATG composites exhibit improved photocatalytic performance toward degradation of methylene blue (MB) and methyl orange (MO) under visible light compared to bare Ag3PO4 and Ag3PO4/TiO2/rGO (ATG). The underlying mechanism has been studied based on the results of reactive oxygen species capture experiment, photoluminescence (PL) spectra, and photocurrent measurements under visible light and monochromatic lights. The improved photocatalytic performance is mainly ascribed to the efficient spatial separation of photo-induced electrons and holes in Ag-ATG, i.e., the electrons in Ag3PO4 transfer to Ag-rGO, meanwhile the holes in Ag3PO4 transfer to TiO2. Ag NPs play an important role in the hybrid structure owing to the synergistic effect of Ag NPs and rGO, which not only enhance the light harvest but also increase the capacity of electron accepting from Ag3PO4. Meanwhile, active photo-induced electrons at the plasmonic Ag NPs can facilitate the formation of O2˙− radicals for photocatalysis. As a result, both the stability and photocatalytic active of Ag-ATG are significantly improved.
Introduction
Visible-light-active semiconductor photocatalysts have attracted widespread attention in recent years due to their great potential for resolving the growing energy and environmental concerns by using solar energy, such as splitting water into H2 and O2,1–3 photoreducing CO2 into renewable fuels (e.g., CH3OH, CH4 and CO)4,5 and decomposing various organic contaminations.6 To date, numerous semiconductor materials, such as Ag-based photocatalysts,7–10 BiOX (X = Cl, Br, I),11,12 Bi2WO6,13 BiVO4,14 C3N4 (ref. 2 and 15) and TiO2−xNx16,17 have been developed as novel visible-light-active photocatalysts. However, how to improve the photocatalytic efficiency is still a great challenge to meet the practical application because the effectiveness of the photocatalytic processes is dictated to a great extent by the semiconductor capability of visible light absorption as well as its ability to suppress the rapid recombination of photo-generated electrons and holes.
Ag3PO4 is a promising visible-light-active photocatalyst due to its extremely high efficiency in water splitting and organic molecules decomposition.7,18 However, the wide application of Ag3PO4 is restricted by the inherent drawbacks, including the high recombination rate of photo-generated electron–hole pairs, the high cost of Ag-containing chemicals as well as the serious photo-corrosion in photocatalysis process. Some modifications of Ag3PO4, such as facet engineering,19,20 morphology and size controlling21,22 can further improve its photocatalytic property, but these modifications cannot resolve all the mentioned drawbacks. In recent years, it has been recognized that the formation of heterostructural composite of two or more semiconductors with matched electronic band structures can promote efficient electron–hole separation and transfer and thus improve the photocatalytic performance.3,23,24 At the same time, the consumption of Ag can be greatly reduced in the composites. Accordingly, many Ag3PO4-based heterostructures, such as Ag3PO4/TiO2,25–27 Ag3PO4/CeO2,28 Ag3PO4/ZnO,29 Ag3PO4/SnO2,30 Ag3PO4/SrTiO3,31,32 Ag3PO4/Bi2MoO6,33 AgBr/Ag3PO4,34 Ag3PO4/WO3,35 Ag3PO4/Sr2Nb2O7,36 BiOCl/Ag3PO4,37 Ag3PO4/carbon nanotubes,38 Ag3PO4/carbon quantum dots,39 Ag3PO4/GO40 and Ag3PO4/rGO41–43 have been developed to improve the stability and activity of Ag3PO4. In the above binary heterostructures, the additional semiconductor (or carbonous material) acts as holes (or electrons) acceptor. For example, in Ag3PO4/TiO2, photo-generated holes in Ag3PO4 can be transferred to TiO2 due to the alignments of the electronic band structures of Ag3PO4 and TiO2,25,26 while in Ag3PO4/rGO, photo-generated electrons in Ag3PO4 can be quickly transferred to rGO sheets.42–44 However, only one pathway, either an electron pathway or a hole pathway, is established in these binary-component photocatalysts. It thus of great interesting to design triple-component composites with both electron and hole pathways, which may further enhance the photo-generated charges separation and result in better photocatalytic performance than the binary-component photocatalysts. Recently, Yang et al. and our group reported the studies of TiO2/Ag3PO4/rGO45 and TiO2/Ag3PO4/GO,46,47 which indeed exhibit superior photocatalytic active and stability compared to binary-component composites. However, the triple-component composites still remain high internal charges recombination due to the relatively large Ag3PO4 particles (200 nm to 2 μm) as well as the insufficient electron/hole transfer in the heterostructure. Therefore, it is desirable to carry out a rational design on the triple-composites that combines the optimization of charges generation, separation and transfer, aiming to develop new Ag3PO4-based photocatalysts with high charges separation efficiency, good stability and low consumption of Ag.
In this paper, we report the construction of a novel Ag-ATG heterostructural composite that possesses superior photocatalytic activity and stability. In comparison with the previously reported triple-component composites,45–47 nano-sized Ag3PO4 immobilized on mesoporous TiO2 spheres not only prevent aggregation of Ag3PO4 but also significantly suppress the bulk charges recombination in Ag3PO4. Ag NPs decorated on rGO sheets play an important role in the new heterostructure, because they can significantly increase the electrical conductivity of rGO that largely facilitates the electrons transfer from Ag3PO4 to Ag-rGO, enhance the visible-light harvest due to the localized surface plasmon resonance (LSPR) effect, and act as the active sites for photocatalytic reaction. The microstructure of the composites and the underlying mechanism for the enhanced photocatalytic performance has been investigated in details.
Experimental section
Materials
All the chemicals were of analytical grade and used without further purification. Natural graphite, methylene blue (MB) and methyl orange (MO) were purchased from Fisher Scientific (Hong Kong) Ltd. Co., Sodium nitrate (NaNO3), potassium permanganate (KMnO4), hydrogen peroxide (H2O2), concentrated sulfuric acid (H2SO4), hydrochloric acid (HCl), silver nitrate (AgNO3), sodium phosphate dibasic dodecahydrate (Na2HPO4·12H2O), sodium phosphate tribasic (Na3PO4), titanium sulfate [Ti(SO4)2], isopropanol (IPA), disodium ethylenediaminetetraacetate (Na2-EDTA) and p-benzoquinone (BQ) were purchased from Sigma-Aldrich. The deionized water was produced from a Millipore Milli-Q water purification system and used throughout the whole experiment.
Synthesis of Ag3PO4/TiO2/Ag-rGO
Fig. 1 illustrates the synthesis process of Ag3PO4/TiO2/Ag-rGO (Ag-ATG) photocatalyst. Ag3PO4/TiO2/rGO (ATG) composite without Ag NPs were also prepared for comparison with Ag-ATG. The photographs of the corresponding intermediate products are also shown in Fig. 1. Firstly, anatase-TiO2/GO composites were prepared using Ti(SO4)2 and GO aqueous suspension as starting materials. In a typical process, 10 mL of GO suspension (3 g L−1) was dispersed in 150 mL sulfuric acid (1 M) solution by ultrasonic treatment for 30 min. Then, Ti(SO4)2 solution prepared by dissolving 3 g Ti(SO4)2 into 50 mL H2SO4 (1 M) was added into the GO solution drop by drop, followed by stirring for 30 min. Next, the mixture solution was subjected to oil bath at 100 °C for 6 hours. After reaction, the TiO2/GO were collected by centrifugation and washed with deionized water. The percentage of GO in TiO2/GO was estimated to be 3 wt%. Secondly, Ag3PO4/TiO2/GO composite was synthesized by a simple in situ precipitation method. In a typical process, a certain 0.2 M Na2HPO4 aqueous solution was added into TiO2/GO solution drop by drop. After ultrasonic treatment for 30 min, a certain 0.02 M AgNO3 aqueous solution was added drop by drop into the above suspension with vigorous stirring in dark condition. After reaction, the brown gray Ag3PO4/TiO2/GO precipitate was collected by centrifugation and washed with deionized water and alcohol for several times. The precipitate was dried in a vacuum oven at 60 °C for 2 h. Finally, Ag3PO4/TiO2/GO was subjected to two different photo-assisted reduction treatments44 for the synthesis of Ag-ATG and ATG composites. For the synthesis of Ag-ATG, Ag3PO4/TiO2/GO was first dispersed into ethanol by ultrasonication. Then, 0.1 M NaOH aqueous solution was added into the Ag3PO4/TiO2/GO solution until the PH reached to 9. Next, a certain 10 mM AgNO3 was added to the solution, followed by stirring for 30 min. After that, the above solution was exposed to UV light irradiation for 5 min.44,48 High purity nitrogen was bubbled through the solution during the UV light irradiation. Finally, Ag-ATG precipitate was collected by centrifugation and washed with deionized water and alcohol for several times, and dried in a vacuum oven at 60 °C for 12 h. ATG sample was prepared under the same condition without adding of AgNO3. In ATG and Ag-ATG photocatalysts, the weight ratio of Ag3PO4 against TiO2/rGO (TG) was controlled to be 0.4, 0.6, 0.8, and 1.0 by adjusting the amount of AgNO3 and Na2HPO4 during the synthesis. The resulting ATG and Ag-ATG samples were labeled as ATG-0.4, ATG-0.6, ATG-0.8, ATG-1.0, Ag-ATG-0.4, Ag-ATG-0.6, Ag-ATG-0.8, Ag-ATG-1.0, respectively. TG prepared by exposed TiO2/GO under UV irradiation was also prepared as a reference sample.
 |
| | Fig. 1 The synthesis process of Ag3PO4/TiO2/rGO (ATG) and Ag3PO4/TiO2/Ag-rGO (Ag-ATG). | |
Characterizations
The phase structures of the samples were characterized by X-ray diffraction (XRD) on a Bruker D8 diffractometer using Cu Kα (λ = 1.5406 Å) radiation at 40 kV and 40 mA. Fourier transform infrared (FTIR) spectra were recorded on a Nicolet Avatar 370 spectrometer with powder samples embedded in KBr disks. Raman spectra were measured by a Bruker Raman microspectrometer with a 532 nm laser. Scanning electron microscopy (SEM) was taken on a Hitachi S-4800 field emission scanning electron microscope at a voltage of 20 kV. Transmission electron microscopy (TEM) images were conducted using a JEM-2100 microscope at an accelerating voltage of 200 kV. The X-ray photoelectron spectra (XPS) were measured using a Kratos Axis Ultra system (Kratos Analytical Ltd., Manchester, UK) with monochromatic Al Kα X-rays (1486.6 eV) operated at 10 mA and 15 kV, and with a background pressure of approximately 5.0 × 10−9 Torr. The binding energies were calibrated to the C 1s (284.6 eV) peak. The UV-Vis diffuse reflectance spectra (UVDRS) were obtained on a Hitachi U3900 UV-Vis spectrophotometer by using BaSO4 as the matrix. The photoluminescence (PL) spectra were obtained on a Hitachi F7000 fluorescence spectrophotometer at an excitation wavelength of 290 nm. Nitrogen adsorption–desorption isotherm measurements were conducted at 77 K (F-sorb 3400).
Photocatalytic performance measurements
The photocatalytic performance of the samples was evaluated by degradation of methylene blue (MB) and methyl orange (MO) in a quartz reactor system. A 300 W Xe lamp equipped with an UV cut filter (λ > 400 nm) was used as the visible-light source. In a typical process, 25 mg photocatalysts was added into 100 mL MB (C0 = 8 mg L−1) or MO (C0 = 10 mg L−1) aqueous solution. Prior to light illumination, the suspension was magnetically stirred for 30 min in dark for attaining adsorption–desorption equilibrium. During illumination, a circulation of water through an external cooling coil was conducted to maintain the temperature of suspension at about 25 °C. At given time intervals of visible-light irradiation, 3 mL of the suspension was collected and centrifugated at 4000 rpm for 15 min, then using a Hitachi U-3900 spectrophotometer to determine the concentration of MB or MO by measuring the absorbance at the maximum peak (λMB,max = 663 nm, λMO,max = 464 nm). To explore the mechanism of the photocatalytic degradation of persistent organic pollutants, IPA (2 mM), Na2-EDTA (2 mM) and BQ (2 mM) acting as ˙OH radical, hole (h+) and O2˙− radical scavengers were added in the photodegradation experiments, respectively. In addition, we repeated the photocatalytic degradation tests for four times in cyclic experiments to examine the stability of Ag3PO4, ATG and Ag-ATG photocatalyts.
Photoelectrochemical measurements
Photoelectrochemical measurements were performed in a conventional three electrode cell, using a Pt plate and an Ag/AgCl electrode (3 M KCl) as counter electrode and reference electrode, respectively. The working electrodes were prepared by the doctor-blade method on fluorine-doped SnO2 (FTO) conductive glass. 10 mg photocatalyst powder was mixed with 2 mL ethanol and 2 μL of Nafion solution (5 wt%), under ultrasonication for 30 min to form slurry. The slurry was then coated onto FTO glass, whose side part was previously protected using Scotch tape, controlling the exposed area of the electrode to be 1 cm2. Here, Nafion solution was used as adhesive to caste powder on the FTO glass.44 After air drying, the electrode was heating at 200 °C for 2 h in vacuum to improve adhesion. The electrolyte was a 0.02 M Na3PO4 aqueous solution degassed with N2 for 2 h prior to the measurements. The transient photocurrent was recorded at a bias voltage of 0.5 V vs. Ag/AgCl. The illumination source was a 150 W halogen lamp equipped with an UV cut filter (λ > 400 nm). The wavelength dependence of photocurrent was also measured with a three-electrode cell under the irradiation of a tunable monochromatic light source (300–600 nm) and at a bias voltage of 0.5 V vs. Ag/AgCl. The monochromatic light was produced by Zolix solar cell QE/IPCE Measurement System (Zolix solar cell scan 100).
Results and discussion
Microstructure characterizations
Fig. 2 shows XRD patterns of TG, Ag3PO4, ATG (sample ATG-0.6) and Ag-ATG (sample Ag-ATG-0.6). The XRD pattern of TG matches the diffraction pattern of anatase TiO2 (JCPDS card no. 21-1272). No diffraction peaks related to rGO are observed in the XRD pattern of TG due to the low amount of rGO and the destruction of regular stacking of graphene sheets by the incorporation of TiO2 particles.39,40 In the XRD pattern of Ag3PO4, all of the diffraction peaks can be indexed to the body-centered cubic phase of Ag3PO4 (JCPDS card no. 06-0505). ATG and Ag-ATG show distinct diffraction peaks of anatase TiO2 and Ag3PO4. As compared with the XRD pattern of ATG, the additional peaks in the pattern of Ag-ATG at 38.1°, 44.3° and 64.4° match well with the (111), (200) and (220) planes of the face-centered cubic Ag (JCPDS card no. 04-0783), indicating the existence of metallic Ag in the Ag-ATG sample.
 |
| | Fig. 2 XRD patterns of TG, Ag3PO4, ATG, and Ag-ATG. | |
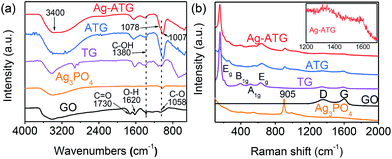
The reduction of GO into rGO in photo-assisted reduction process was investigated with FTIR and Raman spectra. Fig. 3(a) shows the FTIR spectra of GO, Ag3PO4, TG, ATG (sample ATG-0.6) and Ag-ATG (sample Ag-ATG-0.6). In the spectrum of GO, absorption bands at 1730, 1380 and 1058 cm−1 are assigned to the stretching vibrations of carboxyl C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, C–OH and alkoxy C–O, respectively, and the band at 1620 cm−1 is corresponding to the bending mode of O–H groups.49 For pure Ag3PO4, a typical adsorption band at 1007 cm−1 is assigned to molecular vibrations of PO43−.50 In the spectrum of TG, the peaks at 500–700 cm−1 are attributed to Ti–O stretching and Ti–O–Ti bridging stretching vibrations.51 The typical absorption bands related to the vibrations of the PO43−, Ti–O–Ti stretching mode, C–O and C–OH stretching mode are also clearly observed in the spectra of ATG and Ag-ATG. In contrast to the spectrum of GO, the peak intensity of CO and alkoxy C–O decreases significantly in the spectra of TG, ATG and Ag-ATG. This indicates the removal of oxygen-containing functional groups and hence the reduction of GO into rGO. Additionally, the C–O absorption peak in Ag-ATG (1078 cm−1) has been shifted to higher wavenumbers compared to that in GO (1058 cm−1), TG (1058 cm−1) and ATG (1058 cm−1), suggesting the existence of charge interaction between rGO and Ag in the Ag-ATG composite.52 Fig. 3(b) shows the Raman spectra of the samples. Two peaks in the range of 1300–1670 cm−1 are observed in the spectra of GO, TG, ATG and Ag-ATG. They are characteristic Raman peaks (D and G bands) of graphene materials. The D band is assigned to edge or in-plane sp3 defects and disordered carbon, whereas the G band arises from the in-plane vibration of ordered sp2-bonded carbon atoms in a two-dimensional hexagonal lattice.53,54 In contrast to GO, the D band shifts from 1363 cm−1 to 1350 cm−1 and the G band shifts from 1620 cm−1 to 1598 cm−1 in Ag-ATG. The similar shifts of D and G bands are also observed in TG and ATG samples. Furthermore, higher ratio of D/G intensity is also observed in the spectra of TG, ATG and Ag-ATG when compared to that of GO. Both the shift of peak position and the increase of the D/G intensity ratio further indicate that GO has been reduced into rGO after UV-assisted photoreduction.54 In addition to the D and G bands, Raman peaks related to the symmetric stretch of P–O–P bonds (410 and 575 cm−1) and the terminal oxygen bond vibration in phosphate chains (905 cm−1) of Ag3PO4 as well as the Eg (153 cm−1), B1g (400 cm−1), A1g (528 cm−1), Eg (636 cm−1) modes of anatase TiO2 are observed in the ATG and Ag-ATG composites.41,55
O, C–OH and alkoxy C–O, respectively, and the band at 1620 cm−1 is corresponding to the bending mode of O–H groups.49 For pure Ag3PO4, a typical adsorption band at 1007 cm−1 is assigned to molecular vibrations of PO43−.50 In the spectrum of TG, the peaks at 500–700 cm−1 are attributed to Ti–O stretching and Ti–O–Ti bridging stretching vibrations.51 The typical absorption bands related to the vibrations of the PO43−, Ti–O–Ti stretching mode, C–O and C–OH stretching mode are also clearly observed in the spectra of ATG and Ag-ATG. In contrast to the spectrum of GO, the peak intensity of CO and alkoxy C–O decreases significantly in the spectra of TG, ATG and Ag-ATG. This indicates the removal of oxygen-containing functional groups and hence the reduction of GO into rGO. Additionally, the C–O absorption peak in Ag-ATG (1078 cm−1) has been shifted to higher wavenumbers compared to that in GO (1058 cm−1), TG (1058 cm−1) and ATG (1058 cm−1), suggesting the existence of charge interaction between rGO and Ag in the Ag-ATG composite.52 Fig. 3(b) shows the Raman spectra of the samples. Two peaks in the range of 1300–1670 cm−1 are observed in the spectra of GO, TG, ATG and Ag-ATG. They are characteristic Raman peaks (D and G bands) of graphene materials. The D band is assigned to edge or in-plane sp3 defects and disordered carbon, whereas the G band arises from the in-plane vibration of ordered sp2-bonded carbon atoms in a two-dimensional hexagonal lattice.53,54 In contrast to GO, the D band shifts from 1363 cm−1 to 1350 cm−1 and the G band shifts from 1620 cm−1 to 1598 cm−1 in Ag-ATG. The similar shifts of D and G bands are also observed in TG and ATG samples. Furthermore, higher ratio of D/G intensity is also observed in the spectra of TG, ATG and Ag-ATG when compared to that of GO. Both the shift of peak position and the increase of the D/G intensity ratio further indicate that GO has been reduced into rGO after UV-assisted photoreduction.54 In addition to the D and G bands, Raman peaks related to the symmetric stretch of P–O–P bonds (410 and 575 cm−1) and the terminal oxygen bond vibration in phosphate chains (905 cm−1) of Ag3PO4 as well as the Eg (153 cm−1), B1g (400 cm−1), A1g (528 cm−1), Eg (636 cm−1) modes of anatase TiO2 are observed in the ATG and Ag-ATG composites.41,55
 |
| | Fig. 3 (a) FTIR and (b) Raman spectra of GO, Ag3PO4, TG, ATG, and Ag-ATG. The inset figure shows a selected portion of the Raman spectrum of Ag-ATG. | |
XPS measurement was performed to determine the chemical composition and valence state of various species, and the results of ATG (sample ATG-0.6) and Ag-ATG (sample Ag-ATG-0.6) are shown in Fig. S1† and 4. The XPS survey spectra [Fig. S1(a)†] confirm the existence of C, Ag, P, O, and Ti in ATG and Ag-ATG. The high resolution spectrum of C 1s [Fig. 4(a)] can be simply divided into three peaks at around 284.8, 286.2 and 288.9 eV, which are respectively attributed to binding energies of sp2 hybridized non-oxygenated ring carbon atoms (C–C), oxygen-containing epoxy/hydroxyl groups (C–O) and carboxyl (O–CO).56 In comparison with the spectrum of GO [Fig. S1(b)†], the absence of CO peak as well as the significant decrease of C–O and O–CO peaks in ATG and Ag-ATG is ascribed to the reduction of GO into rGO after photo-assisted reduction.44 The shift of C–O binding energy from 286.2 eV (ATG) to 286.7 eV (Ag-ATG) might be ascribed to the combination of Ag and rGO, which significantly increases the electron accepting capacity of rGO.57 The Ag 3d spectra of ATG and Ag-ATG samples are shown in Fig. 4(b). The peaks with binding energies at around 367.8 and 373.8 eV are assigned to Ag 3d3/2 and Ag 3d5/2, indicating the existence of Ag+ in the composites.54 It can be found that the Ag 3d peaks of Ag-ATG shift toward higher binding energy compared to those of ATG. These peak shifts reveal the electronic interaction between Ag and the rGO sheets and the formation of metallic Ag0 in Ag-ATG.57 By fitting the Ag 3d XPS peaks of Ag-ATG [Fig. 4(b)], both peaks can be deconvoluted into two peaks. The strong peaks at 374.1 and 368.1 eV are assigned to Ag+ and the additional weak peaks at 374.5 and 368.6 eV are attributed to metallic Ag0.58 These results agree well with the XRD results, further confirming the formation of metallic Ag0 in the Ag-ATG composite.
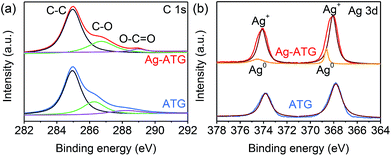 |
| | Fig. 4 XPS spectra of ATG and Ag-ATG: (a) C 1s, (b) Ag 3d. | |
The microstructures of TG, ATG (sample ATG-0.6) and Ag-ATG (sample Ag-ATG-0.6) were investigated by SEM and TEM, as shown in Fig. 5. The SEM and TEM images of TG [Fig. 5(a) and (b)] demonstrate that TiO2 spheres with an average diameter of 200 nm are well wrapped by rGO sheets. From the TEM image [Fig. S2(a)†] of a TiO2 sphere, it is found that the sphere consists of wormhole-like pores with an average size of 4 nm. Nitrogen adsorption–desorption isotherm measurements further confirms the mesoporous structure of TG composite with Brunauer–Emmett–Teller (BET) specific surface area of 87.74 m2 g−1. The clear lattice fringes with a spacing of 0.352 nm in the HRTEM image [Fig. S2(b)†] are assigned to the (101) planes of the anatase TiO2. It is considered that this unique microstructure, containing abundant mesopores, would benefit the immobilization of nano-sized Ag3PO4 on the TiO2 spheres in the subsequent in situ precipitation process. The SEM images in Fig. S3(a) and (b)† show that the ATG and Ag-ATG composites have similar appearance morphology as TG. However, the TEM images of ATG [Fig. 5(c) and (d)] and Ag-ATG [Fig. 5(e)] show absolutely different microstructure from TG. All of the TiO2 spheres in the composites are decorated with small Ag3PO4 NPs (∼10 nm), and only a few Ag3PO4 are dispersed on the rGO sheets. The existence of Ag3PO4 NPs in ATG and Ag-ATG is confirmed by the HRTEM images [Fig. S3(c) and (d)†]. The lattice fringes with a spacing of 0.269 nm is assigned to the (210) planes of Ag3PO4. In contrast to ATG, many nanoparticles with dark contrast are observed on the rGO sheets of the Ag-ATG sample. These nanoparticles are confirmed to be Ag NPs by the HRTEM image in Fig. 5(g). The lattice spacing of 0.235 nm is corresponding to the (111) planes of Ag. The particle-size distribution in Fig. 5(h) deduced from TEM images shows that the Ag NPs fall in the size range of 1–4 nm, and the mean particle diameter is about 2.86 nm. These results agree with the XRD patterns and XPS spectra. Herein, a novel Ag NPs modified rGO wrapping Ag3PO4/TiO2 heterostructural photocatalysts has been constructed.
 |
| | Fig. 5 (a) SEM and (b) TEM images of TG, TEM images of (c, d) ATG and (e–g) Ag-ATG, and (h) size distribution of Ag NPs in Ag-ATG. | |
In the synthesis process, the selective deposition of nano-sized Ag3PO4 and Ag NPs in the composites is controlled via the adding of Ag+ as well as the pH value of the solution. The pH value of the solution can change the surface charge state of the intermediate products and hence result in the selective adsorption of Ag+, which determinate the position of Ag3PO4 and Ag NPs in the composites.44,59,60 Fig. 4S† schematically illustrated the formation mechanism of Ag-ATG. In the step of in situ precipitation, PO43− pre-adsorbing on the positively charged TiO2 spheres of TG react with Ag+ to form Ag3PO4/TiO2/GO at acidic condition (pH = 3).59,60 In the next step, the secondly added Ag+ selectively adsorbed on negatively charged GO surface at a pH value of 9.44 Finally, photo-generated electrons from Ag3PO4 reduce Ag+ and GO into Ag NPs and rGO simultaneously when Ag3PO4/TiO2/GO is exposed to the light irradiation. Ag3PO4 would not be decomposed to Ag during irradiation, because the photo-generated electrons in Ag3PO4 can be quickly transferred to Ag-rGO.42–44
Optical absorption properties
The UVDRS measurements were carried out to study the optical absorption properties of TiO2, Ag3PO4, TG, ATG and Ag-ATG samples. Fig. 6 shows the UVDRS spectra and colors of the samples. Bare TiO2 and Ag3PO4 show sharp absorption edge at about 390 and 530 nm, corresponding to their band-gaps of around 3.20 and 2.34 eV, respectively. TG exhibits stronger absorption in the visible region compared to pure TiO2 due to the incorporation of rGO sheets. Both ATG and Ag-ATG demonstrate stronger absorption than pure Ag3PO4. This can be attributed to the LSPR effect of Ag NPs on rGO and the restoring of the electronic conjugation within the rGO sheets.61 In Ag-ATG, both the Ag NPs and the rGO sheets can absorb photons in visible-light region, so the absorption of Ag-ATG in visible region is higher than that of ATG. The UVDRS spectra of the samples are in agreement with their appearance colors (the inset of Fig. 6). It should be noted that the UVDRS spectral profiles for Ag-ATG with different content of Ag3PO4 are similar except for difference in absorbance intensity. Higher Ag3PO4 content in the composites leads to stronger light absorbance. The strong visible-light absorption for Ag-ATG composite is proposed to generate more electrons and holes for the photocatalytic reaction.
 |
| | Fig. 6 UVDRS spectra of TiO2, Ag3PO4, TG, ATG (ATG-0.6 sample) and Ag-ATG (Ag-ATG-0.6 sample). The inset shows the color of the samples. | |
Photocatalytic performance under visible light
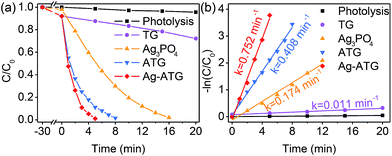
The visible-light-active photocatalytic performance of TG, Ag3PO4, ATG and Ag-ATG composites was evaluated by degradation of negatively charged methyl orange (MO) and positively charged methylene blue (MB), which are hazardous models commonly used to test the degradation capability of photocatalysts.62 Before light irradiation, the adsorption–desorption equilibrium of MO or MB has been established by keeping the solution in dark for 30 min. It is found that TG, ATG and Ag-ATG exhibit slightly higher adsorption property than pure Ag3PO4 [Fig. 7(a) and 8(a)], which is mainly attributed to the high-surface-area rGO that increases the adsorption of organic molecules on the surface of photocatalysts. The temporal evolution of the UV-Vis absorption spectral changes of MO and MB solution mediated by Ag-ATG (sample Ag-ATG-0.6) under visible-light irradiation are shown in Fig. S5,† indicating the degradation of dyes.50 Fig. 7 shows the results of photocatalytic degradation of MO under visible light. The self-photolysis of MO in the absence of photocatalysts can be ignored. All the Ag3PO4-based photocatalysts exhibit prominent photocatalytic activity for MO degradation [Fig. 7(a)], particularly ATG and Ag-ATG composites completely degrade MO within 12 and 7 min, respectively. In contrast, it takes 90 min to decompose 90% MO for pure Ag3PO4 and 10% MO for TG. The photocatalytic activity of the samples follows the order of TG < Ag3PO4 < ATG < Ag-ATG. The apparent rate constants (k) calculated from the degradation curves of −ln(C/C0) versus irradiation time are 0.001, 0.025, 0.244 and 0.510 min−1 for TG, Ag3PO4, ATG and Ag-ATG [Fig. 7(b)], respectively. The k value of Ag-ATG is about 2 times that of ATG and 20 times that of Ag3PO4. The degradation curves of MO by Ag-ATG composites with different contents of Ag3PO4 are given in Fig. 7(c). The photocatalytic activity is dependent on the content of Ag3PO4 in the composites, and the Ag-ATG-0.6 sample exhibits the highest photocatalytic activity. Either a low content or a high content of Ag3PO4 results in relatively low photocatalytic activity. Since Ag3PO4 is main semiconductor to absorb visible light in Ag-ATG composite, it is considered that low content of Ag3PO4 cause weak absorption of visible light. While excess Ag3PO4 in the composite can cause serious aggregation (Fig. S6†) and induce dramatically loss of photo-generated charges due to recombination. In addition to MO, MB was also applied to evaluate the photocatalytic activity of Ag-ATG. Apparently, Ag-ATG exhibits superior photo-degradation activity in MB degradation compared to ATG and Ag3PO4 [Fig. 8(a)]. The degradation activity of MB over the samples follows the same order as that of MO, i.e., TG < Ag3PO4 < ATG < Ag-ATG. The degradation rate constant of Ag-ATG (0.752 min−1) is about 4 times that of Ag3PO4 (0.174 min−1) [Fig. 8(b)].
 |
| | Fig. 7 (a) MO photodegradation as a function of illumination time for Ag3PO4, TG, ATG (ATG-0.6 sample) and Ag-ATG (Ag-ATG-0.6 sample) under visible light (λ > 400 nm); (b) the plots of −ln(C/C0) vs. t; (c) MO photodegradation for Ag-ATG composites with various content of Ag3PO4. C0 is the initial concentration of MO and C is the concentration at irradiation time t. | |
 |
| | Fig. 8 (a) MB photodegradation as a function of illumination time for Ag3PO4, TG, ATG (ATG-0.6 sample) and Ag-ATG (Ag-ATG-0.6 sample) under visible light (λ > 400 nm); (b) the plots of −ln(C/C0) vs. t. C0 is the initial concentration of MB and C is the concentration at irradiation time t. | |
In order to examine the stability of the samples, four runs of cycling photo-degradation MO experiments under the identical condition were carried out for Ag3PO4, ATG and Ag-ATG [Fig. 9]. The degradation rate of MO over bare Ag3PO4 decreases significantly after four runs of cycling photo-degradation experiments. The yellow Ag3PO4 powder became darker during the photocatalytic reaction, indicating serious photo-corrosion. By contrast, both ATG and Ag-ATG maintain high level of photocatalytic activity, and no obvious color changes can be observed on ATG and Ag-ATG composites in the cyclic experiments. The Ag-ATG sample exhibits the highest stability. After four successive cycles, Ag-ATG still gives an almost 100% degradation of MO within 15 min. From the above results, it is concluded that the Ag-ATG composite shows superior photocatalytic performance (including activity and stability), indicating its great potential in practical application.
 |
| | Fig. 9 The plots of cycling photodegradation of MO by Ag3PO4, ATG (sample ATG-0.6) and Ag-ATG (sample Ag-ATG-0.6) under visible light. | |
Mechanism for the improved photocatalytic activity
To explore the mechanism for the improved photocatalytic activity of Ag-ATG, we carried out reactive oxygen species trapping experiments in photocatalytic process. Three different scavengers, disodium ethylenediaminetetraacetate (Na2-EDTA, an h+ scavenger), p-benzoquinone (BQ, an O2˙− radical scavenger) and isopropanol (IPA, a ˙OH radical scavenger) were added in the photocatalytic reaction, respectively. It is found that the photo-degradation of MO by Ag-ATG is significantly suppressed by the introduction of Na2-EDTA, while not obviously suppressed by IPA [Fig. 10]. The photocatalytic activity of Ag-ATG reduces 40% with the adding of BQ. It is concluded that h+ and O2˙− are the main reactive oxygen species involved in the photocatalytic process of Ag-ATG.
 |
| | Fig. 10 Photocatalytic degradation of MO by Ag-ATG under visible light in the presence of different scavengers. | |
PL emission spectra were measured to study the photo-generated charge recombination and transfer behavior of the photocatalysts [Fig. 11]. The emission intensities of the ATG and Ag-ATG composites are obviously suppressed when compared with that of TiO2, TG and Ag3PO4. The significant quenching of the PL emission indicates that the recombination of photo-generated electron–hole pairs in these composites has been effectively suppressed. In the case of ATG, the inhibited charges recombination is explained in terms of the interfacial electron transfer from Ag3PO4 to rGO sheets and hole transfer from Ag3PO4 to TiO2.46 Compared with the PL spectrum of ATG, the even weaker PL emission in Ag-ATG indicates that the Ag NPs can further reduce the recombination rate. This can be ascribed to the improved electrical conductivity of rGO sheets by the decoration of Ag NPs.63 The efficient charge separation in Ag-ATG could extend the lifetime of the photo-generated charges and notably improve the photocatalytic performance.
 |
| | Fig. 11 The PL spectra of TiO2, Ag3PO4, TG, ATG and Ag-ATG under an excitation wavelength of 290 nm. | |
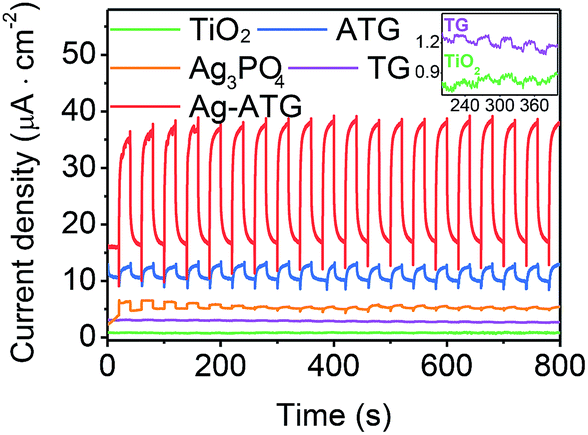
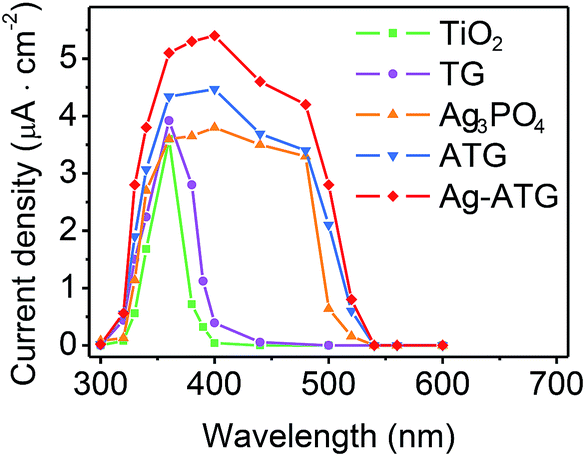
The visible-light-induced photocurrent response of TiO2, Ag3PO4, TG, ATG and Ag-ATG samples were also investigated, and the results [Fig. 12] further confirm the improved charges separation and transfer in Ag-ATG. In Fig. 12, the Ag3PO4 electrode displays photocurrent density of about 1.5 μA cm−2 at the beginning of light irradiation. With the extension of irradiation, the photocurrent density decreases gradually, indicative of the weak stability of Ag3PO4 in the photoelectrochemical reaction. In contrast to TiO2, slight improvement of photocurrent density could be observed in TG (insert of Fig. 12), but the photocurrent density is very small and is almost neglectable. Both the Ag-ATG (sample Ag-ATG-0.6) and ATG (sample ATG-0.6) electrodes show constant photocurrent densities under long time irradiation, indicating the strong stability of ATG and Ag-ATG. It is interesting that the photocurrent density of Ag-ATG (22 μA cm−2) are much higher than that of ATG (4 μA cm−2), indicating that Ag NPs has a strong impact on the charge generation, separation and transfer in the composite. The photocurrent responses of the samples under monochromatic light are also measured. Fig. 13 shows the photocurrent density as a function of light wavelength. TiO2 shows photocurrent response in the ultraviolet region. In contrast, TG exhibits improved photocurrent and broaden photoresponse region due to the incorporation of rGO sheets. Similar to the UVDRS spectrum, Ag3PO4 exhibits photocurrent response in wavelength shorter than 530 nm, matching its band-gap of 2.36 eV. In comparison with bare Ag3PO4, ATG and Ag-ATG have higher photocurrents in the whole wavelength, and the photocurrent responses are slightly red shift to high wavelength. Ag-ATG exhibits the highest photocurrent density and the broadest photoresponse region, which is consistent with the results in Fig. 12. We consider that the improved photocurrent density in Ag-ATG is attributed to the strong light absorption [Fig. 6], the improved charges separation [Fig. 11] as well as the improved electrical conductivity of rGO by Ag NPs modification. The high and stable photocurrent response of Ag-ATG agrees well with its superior photocatalytic performance in MB and MO degradation.
 |
| | Fig. 12 The periodic on/off photocurrent density response of TiO2, TG, Ag3PO4, ATG (sample ATG-0.6) and Ag-ATG (sample Ag-ATG-0.6) electrodes under visible light irradiation (λ > 400 nm) at 0.5 V bias potential vs. Ag/AgCl. The insert is the partial enlarged detail of the TiO2 and TG curves. | |
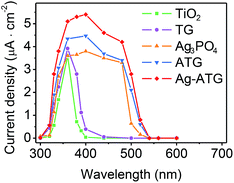 |
| | Fig. 13 The photocurrent density response of TiO2, TG, Ag3PO4, ATG (sample ATG-0.6) and Ag-ATG (sample Ag-ATG-0.6) electrodes under the monochromatic light at 0.5 V bias potential vs. Ag/AgCl. | |
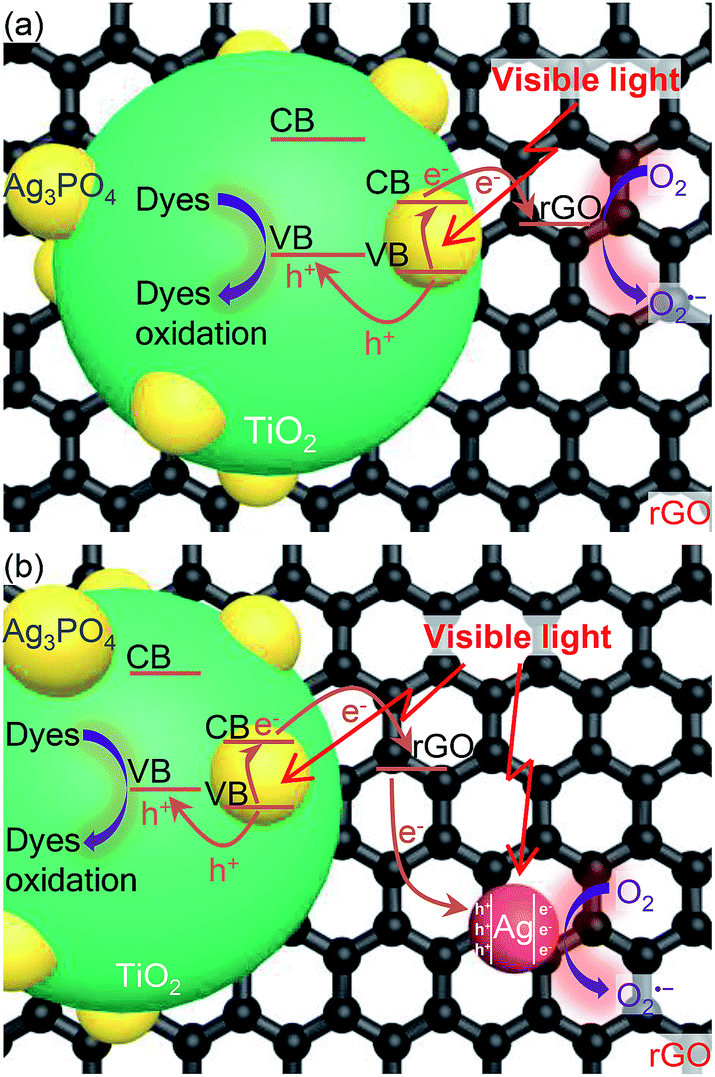
Based on the above experimental results, the photo-induced charges generation, separation and transfer in Ag-ATG are proposed and schematically illustrated in Fig. 14(b) to explain its significantly improved photocatalytic performance. The schematic structure of ATG [Fig. 14(a)] is also illustrated for comparison. Under visible-light irradiation, electrons (e−) are excited to the conduction band (CB) of Ag3PO4, leaving holes (h+) in the valence band (VB). For bare Ag3PO4, normally, these photo-generated e− and h+ undergo rapidly recombination and only a few of them migrate to the surface to take part in the photocatalytic reaction. Some of the electrons could reduce Ag+ in Ag3PO4 into metallic Ag and cause the well-known photo-corrosion phenomenon.7 In ATG and Ag-ATG composites, the novel microstructures as well as the interfacial conjunctions of Ag3PO4/TiO2 and Ag3PO4/rGO (or Ag3PO4/Ag-rGO) improve the charges separation and transfer. Firstly, nano-size Ag3PO4 could shorten the migration distance of photo-generated carriers and favor the charges migration from the bulk to the surface of Ag3PO4. Secondly, the potential of the VB (2.7 eV vs. NHE) of TiO2 are more negative than that of Ag3PO4 (2.81 eV vs. NHE).7,64 Therefore, the h+ tend to aggregate in the VB of TiO2, resulting in an inter-semiconductor transfer channel for h+ (Ag3PO4 → TiO2). Simultaneously, the photo-generated e− in the CB of Ag3PO4 tend to transfer to rGO sheets due to its fine electronic conductivity and the specific π-conjugation structure,64 achieving a transfer channel for e− (Ag3PO4 → rGO). The h+ in the VB of TiO2 could directly oxidized organic molecules, meanwhile the e− accumulated at the rGO (or Ag-rGO) sheets could absorb O2 to form O2˙−, as depicted in Fig. 14(a) and (b). In addition to the above advantages, the Ag NPs dispersed on rGO sheets play a key role in the improved photocatalytic performance of Ag-ATG [Fig. 14(b)]. First, the Ag NPs can improve the electrical conductivity of rGO sheets and results in a high efficient e− transfer from Ag3PO4 to Ag-rGO.65 The fast transfer of e− enhances the stability of Ag3PO4 in the composites. Second, plasmonic Ag NPs can absorb visible light to excite additional e− and h+ due to the LSPR effect.66 The active photo-induced e− in Ag NPs can react with O2 to form O2˙− radicals and leave behind h+ in Ag NPs to form Ag+, which can subsequently capture e− from Ag3PO4 through rGO and regenerate Ag NPs on the surface of rGO.67 Therefore, the photo-excited Ag NPs can promote the formation of O2˙− and provide abundant active sites for photocatalytic reaction. In summary, in comparison with ATG, the accelerated e− transfer and the promoted O2˙− generation by Ag NPs further improve the photocatalytic activity and stability of Ag-ATG.
 |
| | Fig. 14 The proposed mechanism for the photodegradation of organic molecules by (a) ATG and (b) Ag-ATG composites. | |
Conclusions
In summary, a novel Ag-ATG photocatalyst has been constructed with a rational design approach. Ag-ATG exhibits superior visible-light-active photocatalytic performance compared to bare Ag3PO4 and ATG. The improved photocatalytic performance is interpreted as the establishing of dual-channel for charges separation in the heterostructure and the synergistic effect of Ag and rGO. Ag NPs play an important role in the hybrid structure, and they not only enhance the light harvest but also increase the capacity of electron accepting from Ag3PO4. Meanwhile, photo-excited Ag NPs can facilitate the formation of additional O2˙− radicals for photocatalysis. As a result, both the stability and photocatalytic active of Ag-ATG are significantly improved. The results demonstrate that an integrated rational design of the microstructure of multi-component composite can further boost the photocatalytic performance of photocatalyst.
Acknowledgements
This work was supported by the National Undergraduate Training Program for Innovation (201410338003), 521 Talents Project of Zhejiang Sci-Tech University, Xinmiao Undergraduate Student Talents Program of Zhejiang Province (2013R406037, 2015R406028), Open Foundation of Zhejiang Provincial Top Key Academic Discipline of Applied Chemistry, Eco-Dyeing & Finishing Engineering (No. YR2012008), and the Young Researchers Foundation of Zhejiang Provincial Key Laboratory of Fiber Materials and Manufacturing Technology at Zhejiang Sci-Tech University (No. 2015QN01).
Notes and references
- X. Chen, L. Liu, P. Y. Yu and S. S. Mao, Science, 2011, 331, 746–750 CrossRef CAS PubMed.
- J. Liu, Y. Liu, N. Liu, Y. Han, X. Zhang, H. Huang, Y. Lifshitz, S.-T. Lee, J. Zhong and Z. Kang, Science, 2015, 347, 970–974 CrossRef CAS PubMed.
- S. J. A. Moniz, S. A. Shevlin, D. J. Martin, Z.-X. Guo and J. Tang, Energy Environ. Sci., 2015, 8, 731–759 CAS.
- S. N. Habisreutinger, L. Schmidt-Mende and J. K. Stolarczyk, Angew. Chem., Int. Ed., 2013, 52, 7372–7408 CrossRef CAS PubMed.
- Q. Kang, T. Wang, P. Li, L. Liu, K. Chang, M. Li and J. Ye, Angew. Chem., Int. Ed., 2015, 54, 841–845 CrossRef CAS PubMed.
- X.-Y. Wu, H.-X. Qi, J.-J. Ning, J.-F. Wang, Z.-G. Ren and J.-P. Lang, Appl. Catal., B, 2015, 168–169, 98–104 CrossRef CAS.
- Z. Yi, J. Ye, N. Kikugawa, T. Kako, S. Ouyang, H. Stuart-Williams, H. Yang, J. Cao, W. Luo, Z. Li, Y. Liu and R. L. Withers, Nat. Mater., 2010, 9, 559–564 CrossRef CAS PubMed.
- X. Wang, S. Li, H. Yu, J. Yu and S. Liu, Chem.–Eur. J., 2011, 17, 7777–7780 CrossRef CAS PubMed.
- L. Liang, Y. Xu, Y. Lei and H. Liu, Nanoscale, 2014, 6, 3536–3539 RSC.
- C. Yu, G. Li, S. Kumar, K. Yang and R. Jin, Adv. Mater., 2014, 26, 892–898 CrossRef CAS PubMed.
- X. Zhang, Z. Ai, F. Jia and L. Zhang, J. Phys. Chem. C, 2008, 112, 747–753 CAS.
- J. C. Ahern, R. Fairchild, J. S. Thomas, J. Carr and H. H. Patterson, Appl. Catal., B, 2015, 179, 229–238 CrossRef CAS.
- G. Zhang, Z. Hu, M. Sun, Y. Liu, L. Liu, H. Liu, C.-P. Huang, J. Qu and J. Li, Adv. Funct. Mater., 2015, 25, 3726–3734 CrossRef CAS.
- R. Li, F. Zhang, D. Wang, J. Yang, M. Li, J. Zhu, X. Zhou, H. Han and C. Li, Nat. Commun., 2013, 4, 1432 CrossRef PubMed.
- G. Liu, T. Wang, H. Zhang, X. Meng, D. Hao, K. Chang, P. Li, T. Kako and J. Ye, Angew. Chem., Int. Ed., 2015, 54, 13561–13565 CrossRef CAS PubMed.
- X. Chen, Y. B. Lou, A. C. S. Samia, C. Burda and J. L. Gole, Adv. Funct. Mater., 2005, 15, 41–49 CrossRef CAS.
- J. Schneider, M. Matsuoka, M. Takeuchi, J. Zhang, Y. Horiuchi, M. Anpo and D. W. Bahnemann, Chem. Rev., 2014, 114, 9919–9986 CrossRef CAS PubMed.
- D. J. Martin, G. Liu, S. J. A. Moniz, Y. Bi, A. M. Beale, J. Ye and J. Tang, Chem. Soc. Rev., 2015, 44, 7808–7828 RSC.
- Y. Bi, S. Ouyang, N. Umezawa, J. Cao and J. Ye, J. Am. Chem. Soc., 2011, 133, 6490–6492 CrossRef CAS PubMed.
- D. J. Martin, N. Umezawa, X. Chen, J. Ye and J. Tang, Energy Environ. Sci., 2013, 6, 3380–3386 CAS.
- Y. Bi, H. Hu, S. Ouyang, G. Lu, J. Cao and J. Ye, Chem. Commun., 2012, 48, 3748–3750 RSC.
- F. Teng, Z. Liu and A. Zhang, Environ. Sci. Technol., 2015, 49, 9489–9494 CrossRef CAS PubMed.
- H. Li, Y. Zhou, W. Tu, J. Ye and Z. Zou, Adv. Funct. Mater., 2015, 25, 998–1013 CrossRef CAS.
- J. L. White, M. F. Baruch, J. E. Pander Iii, Y. Hu, I. C. Fortmeyer, J. E. Park, T. Zhang, K. Liao, J. Gu, Y. Yan, T. W. Shaw, E. Abelev and A. B. Bocarsly, Chem. Rev., 2015, 115, 12888–12935 CrossRef CAS PubMed.
- W. Yao, B. Zhang, C. Huang, C. Ma, X. Song and Q. Xu, J. Mater. Chem., 2012, 22, 4050–4055 RSC.
- W. Teng, X. Li, Q. Zhao and G. Chen, J. Mater. Chem. A, 2013, 1, 9060–9068 CAS.
- Z. W. Tong, D. Yang, Y. Y. Sun, Y. Tian and Z. Y. Jiang, Phys. Chem. Chem. Phys., 2015, 17, 12199–12206 RSC.
- Z.-M. Yang, G.-F. Huang, W.-Q. Huang, J.-M. Wei, X.-G. Yan, Y.-Y. Liu, C. Jiao, Z. Wan and A. Pan, J. Mater. Chem. A, 2014, 2, 1750–1756 CAS.
- Y.-G. Lin, Y.-K. Hsu, Y.-C. Chen, S.-B. Wang, J. T. Miller, L.-C. Chen and K.-H. Chen, Energy Environ. Sci., 2012, 5, 8917–8922 CAS.
- L. Zhang, H. Zhang, H. Huang, Y. Liu and Z. Kang, New J. Chem., 2012, 36, 1541–1544 RSC.
- X. Guan and L. Guo, ACS Catal., 2014, 4, 3020–3026 CrossRef CAS.
- T. Xian, H. Yang, L. Di, J. Ma, H. Zhang and J. Dai, Nanoscale Res. Lett., 2014, 9, 327 CrossRef PubMed.
- Y.-S. Xu and W.-D. Zhang, Dalton Trans., 2013, 42, 1094–1101 RSC.
- W.-S. Wang, H. Du, R.-X. Wang, T. Wen and A.-W. Xu, Nanoscale, 2013, 5, 3315–3321 RSC.
- Y. Chang, K. Yu, C. Zhang, R. Li, P. Zhao, L.-L. Lou and S. Liu, Appl. Catal., B, 2015, 176–177, 363–373 CrossRef CAS.
- J. Guo, H. Zhou, S. Ouyang, T. Kako and J. Ye, Nanoscale, 2014, 6, 7303–7311 RSC.
- B. Cao, P. Dong, S. Cao and Y. Wang, J. Am. Ceram. Soc., 2013, 96, 544–548 CrossRef CAS.
- Z. Wang, L. Yin, M. Zhang, G. Zhou, H. Fei, H. Shi and H. Dai, J. Mater. Sci., 2014, 49, 1585–1593 CrossRef CAS.
- H. Zhang, H. Huang, H. Ming, H. Li, L. Zhang, Y. Liu and Z. Kang, J. Mater. Chem., 2012, 22, 10501–10506 RSC.
- G. Chen, M. Sun, Q. Wei, Y. Zhang, B. Zhu and B. Du, J. Hazard. Mater., 2013, 244–245, 86–93 CrossRef CAS PubMed.
- P. Dong, Y. Wang, B. Cao, S. Xin, L. Guo, J. Zhang and F. Li, Appl. Catal., B, 2013, 132, 45–53 CrossRef.
- X. Yang, H. Cui, Y. Li, J. Qin, R. Zhang and H. Tang, ACS Catal., 2013, 3, 363–369 CrossRef CAS.
- Q. Xiang, D. Lang, T. Shen and F. Liu, Appl. Catal., B, 2015, 162, 196–203 CrossRef CAS.
- C. Cui, Y. Wang, D. Liang, W. Cui, H. Hu, B. Lu, S. Xu, X. Li, C. Wang and Y. Yang, Appl. Catal., B, 2014, 158–159, 150–160 CrossRef CAS.
- N. Ma, Y. Qiu, Y. Zhang, H. Liu, Y. Yang, J. Wang, X. Li and C. Cui, J. Alloys Compd., 2015, 648, 818–825 CrossRef CAS.
- B. Lu, N. Ma, Y. Wang, Y. Qiu, H. Hu, J. Zhao, D. Liang, S. Xu, X. Li, Z. Zhu and C. Cui, J. Alloys Compd., 2015, 630, 163–171 CrossRef CAS.
- X. Yang, J. Qin, Y. Jiang, K. Chen, X. Yan, D. Zhang, R. Li and H. Tang, Appl. Catal., B, 2015, 166–167, 231–240 CrossRef CAS.
- G. Williams, B. Seger and P. V. Kamat, ACS Nano, 2008, 2, 1487–1491 CrossRef CAS PubMed.
- Y. H. Ding, P. Zhang, Q. Zhuo, H. M. Ren, Z. M. Yang and Y. Jiang, Nanotechnology, 2011, 22, 215601 CrossRef CAS PubMed.
- Q. Liang, W. Ma, Y. Shi, Z. Li and X. Yang, CrystEngComm, 2012, 14, 2966–2973 RSC.
- J.-G. Yu, H.-G. Yu, B. Cheng, X.-J. Zhao, J. C. Yu and W.-K. Ho, J. Phys. Chem. B, 2003, 107, 13871–13879 CrossRef CAS.
- J. Petroski and M. A. El-Sayed, J. Phys. Chem. A, 2003, 107, 8371–8375 CrossRef CAS.
- G. Zhao, L. Jiang, Y. He, J. Li, H. Dong, X. Wang and W. Hu, Adv. Mater., 2011, 23, 3959–3963 CrossRef CAS PubMed.
- Y. Fan, W. Ma, D. Han, S. Gan, X. Dong and L. Niu, Adv. Mater., 2015, 27, 3767–3773 CrossRef CAS PubMed.
- N. Li, G. Liu, C. Zhen, F. Li, L. Zhang and H.-M. Cheng, Adv. Funct. Mater., 2011, 21, 1717–1722 CrossRef CAS.
- Q. Li, B. Guo, J. Yu, J. Ran, B. Zhang, H. Yan and J. R. Gong, J. Am. Chem. Soc., 2011, 133, 10878–10884 CrossRef CAS PubMed.
- S. Dutta, C. Ray, S. Sarkar, M. Pradhan, Y. Negishi and T. Pal, ACS Appl. Mater. Interfaces, 2013, 5, 8724–8732 CAS.
- B. Jiang, Y. Wang, J.-Q. Wang, C. Tian, W. Li, Q. Feng, Q. Pan and H. Fu, ChemCatChem, 2013, 5, 1359–1367 CrossRef CAS.
- R. Zanella, S. Giorgio, C. R. Henry and C. Louis, J. Phys. Chem. B, 2002, 106, 7634–7642 CrossRef CAS.
- X. Guo, N. Chen, C. Feng, Y. Yang, B. Zhang, G. Wang and Z. Zhang, Catal. Commun., 2013, 38, 26–30 CrossRef CAS.
- D. Li, M. B. Muller, S. Gilje, R. B. Kaner and G. G. Wallace, Nat. Nanotechnol., 2008, 3, 101–105 CrossRef CAS PubMed.
- X. Luo and L. Zhang, J. Hazard. Mater., 2009, 171, 340–347 CrossRef CAS PubMed.
- P. Wang, Y. Tang, Z. Dong, Z. Chen and T.-T. Lim, J. Mater. Chem. A, 2013, 1, 4718–4727 CAS.
- S. B. Rawal, S. D. Sung and W. I. Lee, Catal. Commun., 2012, 17, 131–135 CrossRef CAS.
- L. Li, Y. Guo, X. Zhang and Y. Song, J. Mater. Chem. A, 2014, 2, 19095–19101 CAS.
- J. Yu, G. Dai and B. Huang, J. Phys. Chem. C, 2009, 113, 16394–16401 CAS.
- M. Rycenga, C. M. Cobley, J. Zeng, W. Li, C. H. Moran, Q. Zhang, D. Qin and Y. Xia, Chem. Rev., 2011, 111, 3669–3712 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra03420a |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.