
Controlled construction of 3D hierarchical manganese fluoride nanostructures via an oleylamine-assisted solvothermal route with high performance for rechargeable lithium ion batteries†
Abstract
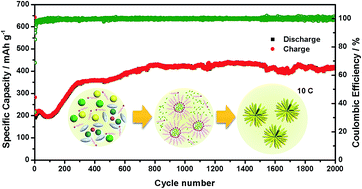
For diverse electrode materials, especially those reacting through a conversion mechanism, nanostructure engineering has proven to be one of the most effective strategies to improve their electrochemical performance. Typically, construction of three-dimensional (3D) nanostructures has offered a valid solution for the challenging issues involved in bulk transition metal based electrode materials for energy storage devices. Herein, we present a facile and effective solvothermal method to fabricate 3D hierarchical dendritic MnF2 nanostructures built from a bunch of radially oriented nanorods. Oleylamine (OAm) is adopted as the surfactant and structural-directing template, giving rise to continuous morphology evolution with different dosages. The electrochemical performance of the as-prepared MnF2 anode for rechargeable lithium batteries is investigated. Long term cycle performance at 10C for 2000 cycles was also obtained with an activated capacity as high as 420 mA h g−1. It is also noteworthy that the activation span was significantly shortened. Desirable MnF2 nanostructures are responsible for the excellent Li-storage capacity with satisfactory cycle performance and superior rate capability.
 Please wait while we load your content...
Please wait while we load your content...

