
Enhancing the biological properties of carbon nanofibers by controlling the crystallization of incorporated bioactive glass via silicon content
Dan Chenga,
Rongrong Xiea,
Le Jina,
Man Caoa,
Xiaolong Jia*ab,
Qing Cai*ac and
Xiaoping Yangabc
aState Key Laboratory of Organic–Inorganic Composites, College of Materials Science and Engineering, Beijing University of Chemical Technology, Beijing 100029, P. R. China. E-mail: jiaxl@mail.buct.edu.cn; caiqing@mail.buct.edu.cn; Fax: +86 10 64412884
bChangzhou Institute of Advanced Materials, Beijing University of Chemical Technology, Jiangsu 213164, P. R. China
cBeijing Laboratory of Biomedical Materials, Beijing University of Chemical Technology, Beijing 100029, P. R. China
First published on 19th May 2016
Abstract
Bioactive glass (BG)-containing carbon nanofibers (CNFs) were prepared by combining the processes of sol–gel, polyacrylonitrile (PAN) electrospinning and heat treatment. Two types of BG, i.e. 45S and 68S, were incorporated. The crystalline structure evolution of the BG component during the formation of the CNFs was characterized by XRD, SEM, and TEM observations in relation to silicon content. Then the apatite-forming ability of the hybridized CNF/BG in simulated body fluid was evaluated in relation to the crystalline structure of the BG component. Interactions between functional groups in PAN and BG sol–gel precursors were identified in the steps of electrospinning and heat treatment. As a result, the 45S-type BG containing less silicon formed α-CaSiO3, while the 68S-type BG containing more silicon transformed to β-CaSiO3 in the final hybridized CNF/BG upon carbonization. This difference led to different dissolution rates and osteocompatibility activities of the BG component from the hybrids, which regulated their capacities in inducing apatite deposition, proliferation and osteogenic differentiation of bone mesenchymal stromal cells.
Bone defect repair remains a challenge in clinical therapy, which calls for osteoinductive substrates to achieve desired bone regeneration.1,2 Biological ceramics such as bioactive glass (BG) have been widely used as osteoinductive substrates in bone tissue engineering due their excellent affinity to both bone and cartilage tissues.3,4 From the viewpoint of mimicking the nanofibrous structure of the natural extracellular matrix, fabrication of BG nanofibers is preferred. However, this meets some difficulties, especially in producing long and continuous BG nanofibers due to the intrinsic brittleness of ceramic materials.4,5 Carbon nanofibers (CNFs) were envisioned as an excellent support material for BG component targeting for bone defect repair applications. CNFs are not only close in size to the triple helical collagen fibrils, but also possess good mechanical properties. Besides, CNFs have been shown to support attachment and proliferation of osteoblastic cells.6–8 Therefore, BG-loaded CNFs (CNF/BG) have been studied as promising orthopaedic biomaterials in publications.7–10 The way to produce CNF/BG is normally to apply the BG sol–gel precursor in combination with polyacrylonitrile (PAN) electrospinning, followed by carbonization. In our previous studies,9,11 the effects of sol–gel aging time, carbonization temperature and time on the crystalline structure of the resulting BG nanoparticles and morphology of the resulting CNF/BG hybrids have been systematically reported. Osteocompatibility of CNF/BG hybrids was identified, which showed dependence on aforementioned factors. In view of the content of silicon in BG component playing an important role in regulating osteoblast proliferation and osteogenic differentiation,12 it was interesting to know how CNF/BG hybrids containing BG with different silicon contents would affect their biological properties. Therefore, in this study, two CNF/BG hybrids were prepared by controlling different silicon contents in BG precursors. Biomineralization in simulated body fluid (SBF) was applied to evaluate the biological properties of resulted CNF/BG hybrids in comparison with pure CNFs. The capacity of the substrate in inducing apatite deposition from SBF has been identified valid in evaluating the osteocompatibility of BG substrates because the process closely related to BG dissolution and bone binding ability.13 In regarding to the silicon content in BG precursors, thus, the morphological and micro-structural evolution of CNF/BG hybrids, and their relationship to apatite-forming ability were systematically investigated.
The fabrication of CNF/BG hybrids was conducted following the same process in our previous reports.9–11 Briefly, calcium nitrate tetrahydrate (CN, Aldrich, USA) and as-hydrolyzed triethyl phosphate (TEP, Aldrich, USA) solution were dissolved into tetraethoxysilane (TEOS, Aldrich, USA) solution in N,N-dimethyl formamide (DMF, analytic pure, 99.5%, Tianjin Fine Chemical Co., China) at the feeding ratios listed in Table 1. Two silicon contents, i.e. 45 wt% and 68 wt%, in preparing BG sol–gel precursor solution were chosen. Noticeably, the Ca/P ratio in the present study was set 1.67 because this value was the theoretical Ca/P ratio of stoichiometric hydroxyapatite (HA), which was the main analogue to natural bone minerals.14 The mixture was then stirred at 37 °C for 7 days to generate a homogeneous sol–gel solution. The sol–gel solution was observed with optical microscope (OM, Olympus, Japan) and submitted to sol–gel particle size measurement using laser particle size analyzer (LPSA, 2000, UK). Next, the as-prepared sol–gel solution was added into a 10 wt% PAN (Mw = 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, composed of 93.0 wt% acrylonitrile, 5.3 wt% methylacrylate, and 1.7 wt% itaconic acid, Courtaulds Co, UK) solution in DMF, and continuously stirred at 37 °C overnight. Fibrous composite membranes were made via electrospinning (15 kV, 0.4 m s−1, 15 cm). Subsequently, the membranes were treated in steps of hydrolysis (60 °C, 24 h), stabilization (280 °C, 2 h, air atmosphere) and carbonization (1000 °C, 3 h, N2 atmosphere) to obtain the final CNF/BG hybrids. For comparison, pure CNFs were prepared under a similar condition. Besides, pure BG sol–gel solutions were also electrosprayed and thermally treated as above. For clarity, samples made from pure BG sol–gel were termed as BG-45 and BG-68, respectively, according to their silicon contents. Accordingly, final CNF/BG hybrids containing BG-45 and BG-68 were termed as CNF/BG-45 and CNF/BG-68. While the as-electrospun pure PAN nanofibers and PAN/BG precursor composite nanofibers were termed as NF, NF-45 and NF-68, respectively. After oxidation, they were renamed as ONF, ONF-45 and ONF-68, accordingly. All these samples were comprehensively characterized by scanning electron microscope (SEM, S-250, UK), Fourier transform infrared spectroscope (FT-IR, Nexus670, Nicolet, USA), high resolution transmission electron microscope (HR-TEM, Hitachi H-800, Japan), X-ray diffraction (XRD, Rigaku D/max 2500 VB2+/PC, Japan) analysis and zeta potential measurement (PALS Zeta Potential Analyzer Ver. 5.76, USA).
000, composed of 93.0 wt% acrylonitrile, 5.3 wt% methylacrylate, and 1.7 wt% itaconic acid, Courtaulds Co, UK) solution in DMF, and continuously stirred at 37 °C overnight. Fibrous composite membranes were made via electrospinning (15 kV, 0.4 m s−1, 15 cm). Subsequently, the membranes were treated in steps of hydrolysis (60 °C, 24 h), stabilization (280 °C, 2 h, air atmosphere) and carbonization (1000 °C, 3 h, N2 atmosphere) to obtain the final CNF/BG hybrids. For comparison, pure CNFs were prepared under a similar condition. Besides, pure BG sol–gel solutions were also electrosprayed and thermally treated as above. For clarity, samples made from pure BG sol–gel were termed as BG-45 and BG-68, respectively, according to their silicon contents. Accordingly, final CNF/BG hybrids containing BG-45 and BG-68 were termed as CNF/BG-45 and CNF/BG-68. While the as-electrospun pure PAN nanofibers and PAN/BG precursor composite nanofibers were termed as NF, NF-45 and NF-68, respectively. After oxidation, they were renamed as ONF, ONF-45 and ONF-68, accordingly. All these samples were comprehensively characterized by scanning electron microscope (SEM, S-250, UK), Fourier transform infrared spectroscope (FT-IR, Nexus670, Nicolet, USA), high resolution transmission electron microscope (HR-TEM, Hitachi H-800, Japan), X-ray diffraction (XRD, Rigaku D/max 2500 VB2+/PC, Japan) analysis and zeta potential measurement (PALS Zeta Potential Analyzer Ver. 5.76, USA).
| Sample | Ca(NO3)2 (g) | Hydrolyzed-TEP (mL) | TEOS (mL) | PAN (g) | DMF (mL) |
|---|---|---|---|---|---|
| CNF/BG-45 | 0.481 | 0.501 | 0.483 | 2 | 20 |
| CNF/BG-68 | 0.268 | 0.269 | 0.761 | 2 | 20 |
To perform the biomineralization, pure CNF and the two CNF/BG hybrids were soaked in SBF. Referring to ISO standard 23317:2007, the SBF was prepared by precisely weighing and dissolving the following reagents into 1 L deionized water: NaCl 11.9925 g, NaHCO3 0.5295 g, KCl 0.3360 g, K2HPO4·3H2O 0.3420 g, MgCl2·6H2O 0.4575 g, CaCl 0.4168 g, Na2SO4 0.1065 g. The solution pH was adjusted to 7.4 at 37 ± 0.2 °C with the buffer solution (0.05 mol L−1) composed of tris(hydroxymethyl)aminomethane (Tris) and hydrochloric acid. Fibrous membranes were cut into rectangular pieces (2 cm × 4 cm) and immersed in the SBF at 37 ± 0.2 °C for 1–7 days. At each predetermined time point, samples were retrieved and gently rinsed with deionized water, and then dried at 80 °C for 24 h. The mineral depositions on pure CNF and CNF/BG hybrids were also analyzed by SEM, FTIR and XRD in relation to the features of substrates.
For biological evaluations, bone mesenchymal stromal cells (BMSCs) were cultured on CNF/BG hybrids to determine cell proliferation and alkaline phosphatase (ALP) activity, using pure CNF and tissue culture polystyrene (TCPS) plate as the controls. Briefly, BMSCs were cultured in Dulbecco's Modified Eagle's Medium (DMEM, Hyclone) supplemented with 10% fetal bovine serum (FBS, Gibco, USA), 100 IU mL−1 penicillin (Sigma), and 100 mg mL−1 streptomycin (Sigma) in an incubator (Sanyo, Japan) with 5% CO2 and saturated humidity at 37 °C. Once the cells reached 80% confluence, the BMSCs were digested by 0.25% trypsin (Sigma) and 0.02% ethylene diamine tetraacetic acid (EDTA) for further use. For osteogenic differentiation assay, 0.05 mmol L−1 vitamin C (Sigma, USA), 10 mmol L−1 β-sodium glycerophosphate (Sigma, USA) and 1 × 10−8 mol L−1 dexamethasone (Sigma, USA) were added to the culture medium. The culture medium and the osteogenic inductive medium were refreshed every two days. The CNF/BG hybrids and pure CNF used for cell culture were cut into circular patches and placed into culture plates. Before cells were seeded, the substrates were immersed in 75% ethanol with exposure to ultraviolet (UV) light for 2 h, followed by three times phosphate buffered saline (PBS) washing and being immersed in α-MEM overnight. Then BMSCs were added into each well and cultured 1–7 days to evaluate cell proliferation by using Cell Counting Kit-8 (CCK-8, Beyotime, China). For ALP activity analysis, at the 3, 7 and 14 days of being induced osteogenic differentiation, cellular constructs were retrieved and rinsed with PBS three times. Cell lysates were obtained by adding 1% Triton X-100 and ALP activity was determined using ALP ELISA kits (Cloud-Clone, USA) following manufacturers' protocols. The ALP activities were normalized to the total protein content determined using the BCA assay kit (Thermo, USA). Three independent experiments for each material were performed for averaging.
The structural transformations of BG-45 and BG-68 sol–gel precursors are shown in Fig. 1. From Fig. 1(a1 and b1), the images taken by OM clearly visualized the particular morphology and size in the precursor solutions of BG-45 and BG-68. In both systems, highly cross-linked sol–gel particles were observed in high density. The BG-45 sol–gel particles with smaller size were much branched and dispersed in comparison with the case of BG-68 sol–gel particles. After being diluted in ethanol, the sizes of sol–gel precursor particles were measured to be 11.75 μm for BG-45 and 16.71 μm for BG-68 using LPSA (Fig. 1(a2 and b2)). The particle size here was the dynamic dimension, which could be considered as the measurement of the movement territory of particles in the precursor solution.15 Then the two-dimensional fractal dimension D2 was applied to describe the situations quantitatively (Fig. 1(a3 and b3)). Referring to both literature16 and our previous reports,17 D2 was obtained by the slop measured through the relationship between areas (A) and perimeters (L) by the definition of A ∝ LD2, where A and L are the area and the perimeter of objects (here are the sol–gel precursor particles). Based on Fig. 1(a1 and b1), logA was plotted versus logL as shown in Fig. 1(a3 and b3). The D2 value of BG-45 sol–gel was calculated to be 1.73, while it was identified to be 1.82 for BG-68 sol–gel. The fractal dimension was an evaluation of the efficiency of space filling of the aggregate structures, and thus the D2 value of two-dimensional objects with regular circular shape was 2 theoretically. Lower D2 value meant the formation of irregular-shaped aggregates with loosely structure, corresponding to larger area proportion of aggregates in unit area. As indicated by the D2 values of BG-45 and BG-68 sol–gel, the former obviously had more loosely-structured sol–gel precursor particles in irregular shapes, which were revealed clearly by the OM observation (Fig. 1(a1 and b1)). The difference in the sol–gel precursor solutions of BG-45 and BG-68 was supposed related to their structural evolution along with the formation of Si–O–Si network, which was apparently dependent on silicon content in the system. CP/MAS 29Si nuclear magnetic resonance (NMR, Bruker, Switzerland) was adopted as a powerful tool to investigate structural changes of Qn for silica-based glasses.18 Qn (n ranging from 0 to 4) stood for silicon atoms in silicate network that were connected to other silicon atoms by n bridging oxygen atoms ascribing to the structures of Si(OSi)n(OR)4−n.19 Therefore, deconvoluted 29Si NMR spectra were shown in Fig. 1(a4 and b4) for BG-45 sol–gel and BG-68 sol–gel, respectively. Integral areas of different types of Qn (n = 2, 3, 4) were identified and listed in Fig. 1(c). It could be seen that the values of QSi3 and QSi4 for BG-45 sol–gel were lower than those for BG-68 sol–gel, illustrating the formation of three-dimensional crosslinked Si-network being slower in the case of BG-45 than in the case of BG-68. According to literature,20 the condensation rate (Dc) could be calculated by using the eqn: Dc = (1 × Q1 + 2 × Q2 + 3 × Q3 + 4 × Q4)/4. Basing on those data presented in Fig. 1(c), the Dc value of BG-45 sol–gel was calculated 74.48%, which was lower than the 76.48% in BG-68 sol–gel. After being aged for 7 days, thus, the Si-network in BG-45 sol–gel was crosslinked to a lower extend than the Si-network in BG-68 sol–gel. The former displayed looser sol–gel particles than the latter, which were consistent with the fact that the D2 value of BG-45 sol–gel precursor particles was lower than that of BG-68 sol–gel precursor particles. It was suggested that less Si–OH groups should be generated upon the hydrolysis of TEOS if the feeding dose of silicon was lower, therefore, it should need longer time to complete the dehydrate-condensing reaction between Si–OH groups.
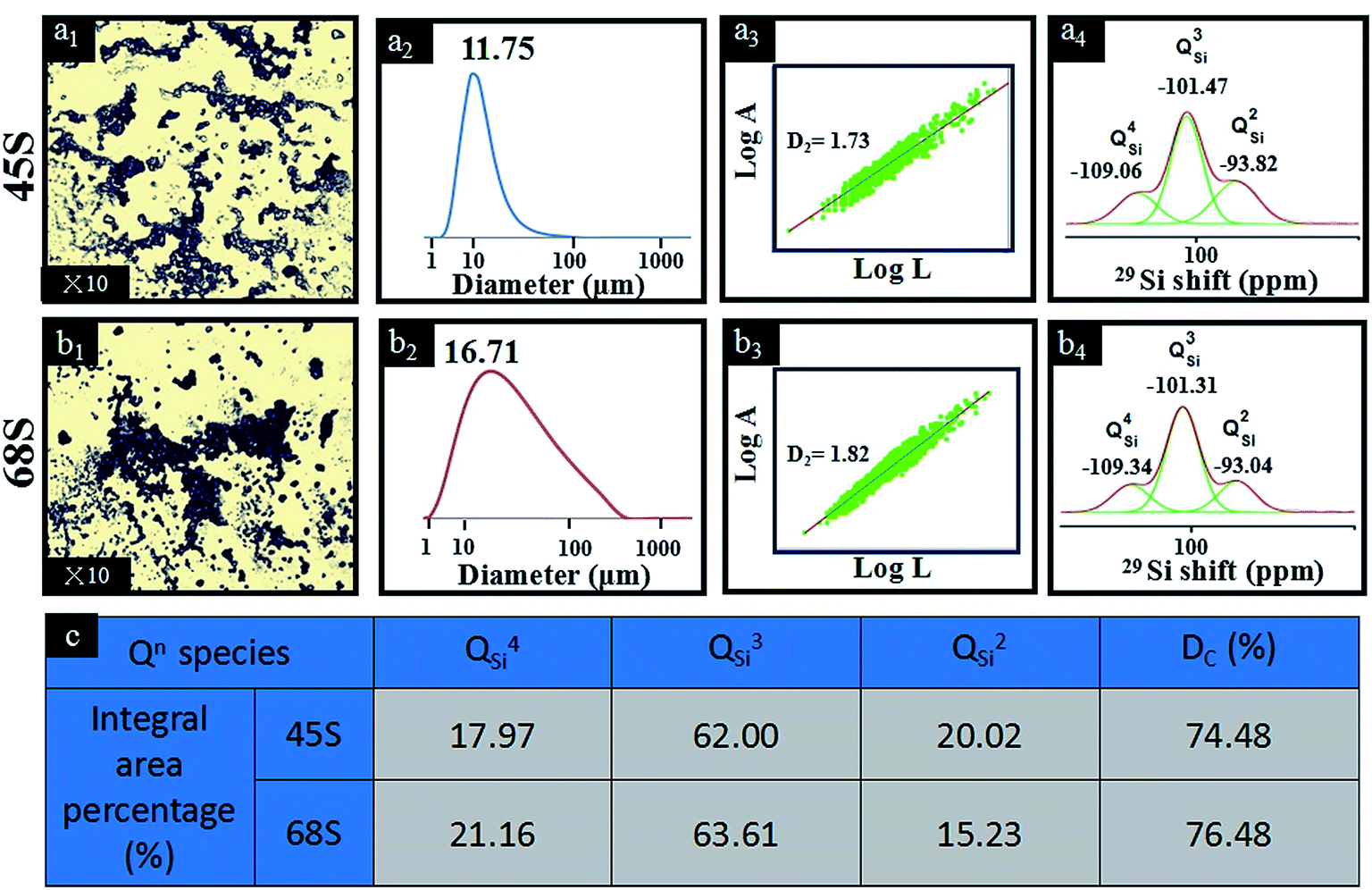 | ||
| Fig. 1 (a1 and b1) Optical microscope images, (a2 and b2) size distribution, (a3 and b3) two-dimensional fractal dimensions (D2), (a4 and b4) deconvoluted 29Si NMR spectra and (c) calculated characteristic parameters of 29Si NMR spectra for BG sol–gel precursors deriving from different silicon contents. In panel (a3) and (b3), the linear fit for the experimental relationship curves of logA versus logL was acquired when correlation coefficient R > 0.97, and the slopes of the curves are D2 values of precursor particles. | ||
An integrated view of the morphology and micro-structure of CNF/BG-45 and CNF/BG-68 hybrids are illustrated in Fig. 2. CNFs in both cases could be seen fully covered by evenly distributed spherical particles (Fig. 2(a1 and b1)). The average particle size in CNF/BG-68 was measured 55.1 nm, which was larger than the average size (19.3 nm) of those particles in CNF/BG-45. In both cases, the spherical particles were identified BG nanoparticles according to our previous reports,9,11 and the difference in their sizes was thought deriving from their sol–gel precursor particles. Both the samples were then submitted to XRD analysis and HR-TEM observation. As shown in Fig. 2(a2 and b2), two broad diffraction peaks at 24.5° and 44.5° were detected for the two samples, which were identified as (0 0 2) and (1 0 1) reflections of graphitic carbon to indicate the formation of CNFs.21,22 Whereas no other peak was detected to show the presence of BG component. It was suggested that the CNF might shield the BG signals in XRD analysis because the content of BG component in CNF/BG hybrid was not quite high.9 As further evaluated by HR-TEM, BG nanoparticles in both CNF/BG-45 and CNF/BG-68 were detected clear lattice fringes in HR-TEM images, demonstrating specific diffraction mode in fast Fourier transform (FFT) patterns (Fig. 2(a3 and b3)). The lattice fringes of d-spacing of 0.245 nm in sample of CNF/BG-45 were assigned to the (1 0 2) plane of α-CaSiO3 according to JCPD card of 01-0720, which was consistent to its diffraction mode in the FFT pattern (Fig. 2(a3)). Differently, the d-spacing value of lattice fringes in sample of CNF/BG-68 was 0.234 nm, which was ascribed to the (−3 −3 2) plane of β-CaSiO3 (JCPD: 84-0654) and confirmed by the FFT pattern (Fig. 2(b3)). Clearly, the BG nanoparticles in both CNF/BG-45 and CNF/BG-68 were crystalline, but in different crystalline structures, which depended apparently on the silicon content in BG sol–gel precursor. In comparing the XRD and HR-TEM results, however, question was raised. Was there any interaction between the BG sol–gel precursor and the PAN precursor of CNF in DMF solution? Would the carbonization in absence of oxygen have effect on formation of crystalline BG nanoparticles?
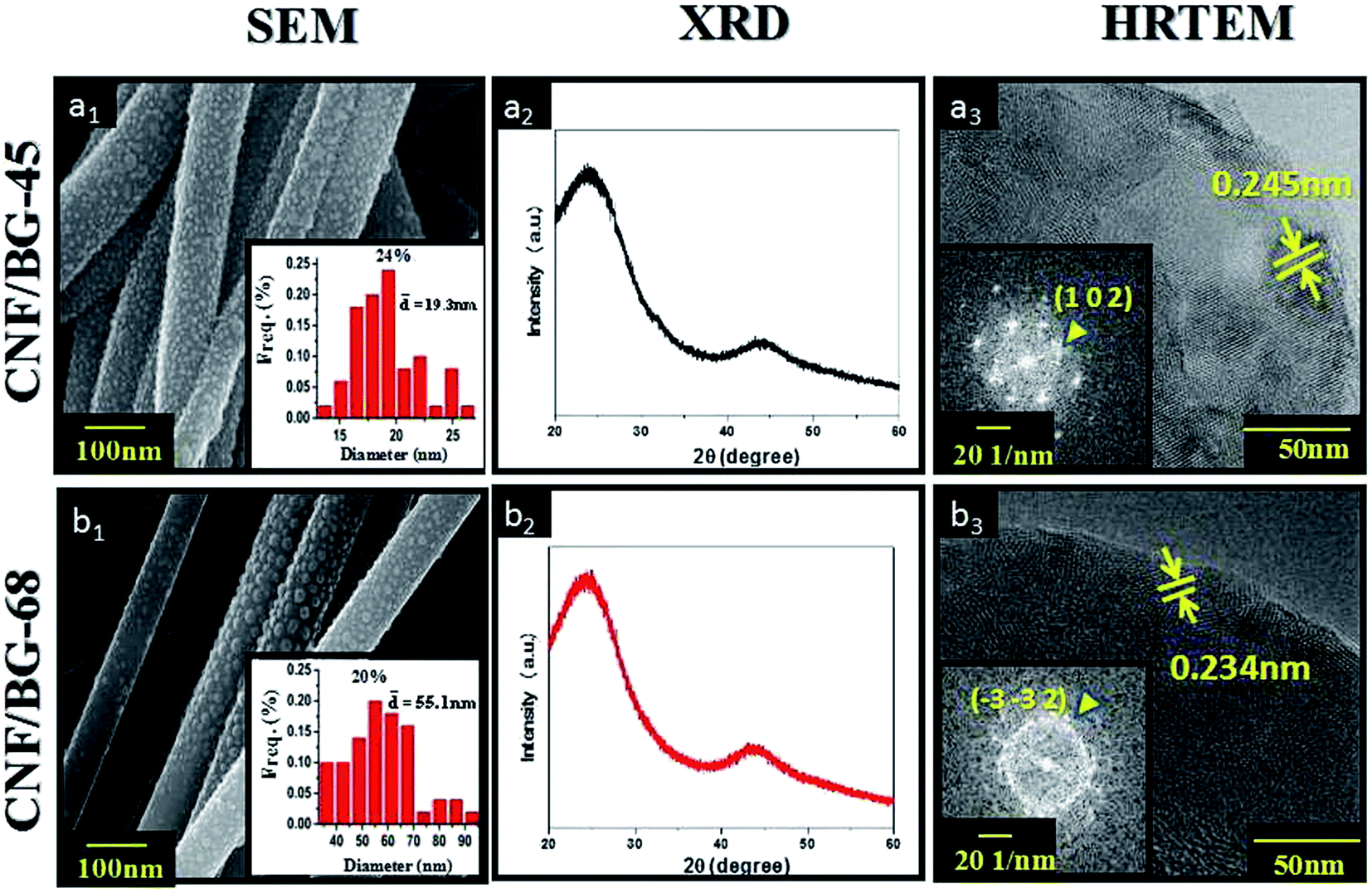 | ||
| Fig. 2 (a1 and b1) SEM images and (a2 and b2) XRD patterns of CNF/BG hybrids, as well as (a3 and b3) HRTEM images of BG nanoparticles in CNF/BG hybrids. The insets in the first and the third columns are the corresponding size distribution images and FFT images of BG nanoparticles in CNF/BG hybrids, respectively. | ||
To answer these questions, detailed studies on sintering were carried out by thermally treating (i) electrosprayed BG sol–gel precursor (BG-45 and BG-68) in nitrogen (1000 °C, 3 h), (ii) electrospun PAN/BG sol–gel composite nanofibers (PAN/BG-45 and PAN/BG-68) in air (1000 °C, 3 h) and (iii) PAN/BG-45 and PAN/BG-68 in nitrogen (1000 °C, 3 h) and air (1000 °C, 3 h) sequentially. Instead of α-CaSiO3 or β-CaSiO3 being detected, the XRD patterns of pure BG-45 and BG-68 resulting from calcination in nitrogen displayed obvious characteristic peaks of HA (Fig. 3(a)), which matched well with JCPD card 09-0432 in Jade software.23–25 Besides, the peak at 22.16° was assigned to cristobalite (SiO2, JCPD: 39-1425),26 and its intensity was higher in the case of BG-68 than in the case of BG-45. The formation of HA phase was suggested being resulted from the feeding Ca/P ratio (1.67), which was similar to that in stoichiometric HA.27,28 The appearance of SiO2 was thought related to the abundant silicon component. However, these results in the absence of PAN were totally different from those results in the presence of PAN, even other parameters were remained consistent. When PAN/BG-45 and PAN/BG-68 were sintered in air instead of nitrogen, no signal of HA was able to be found in XRD anymore (Fig. 3(b)). In this situation, only inorganic components would remain due to the decomposition and the oxidation of organic components during the sintering in air. And the resulted powders in both cases demonstrated the formation of β-CaSiO3. In comparison with Fig. 3(a), it was inferred that the presence of PAN component indeed had influence on crystallization of BG component. Thus, PAN/BG-45 and PAN/BG-68 were carbonized in nitrogen initially and then sintered in air to remove the carbon component. Interestingly, the XRD patterns of resulted BG components still differed from the previous two situations. As shown in Fig. 3(c), the peaks at 26.04°, 27.78°, 32.09° and 45.78° in sample of BG-45 were assigned to α-CaSiO3,29 while the peaks at 27.34°, 29.82°, 31.65° and 49.66° in sample of BG-68 were assigned to β-CaSiO3.30–32 This finding, however, was consistent with those results of HR-TEM and FFT patterns (Fig. 2(a3 and b3)). During the carbonization in nitrogen, apparently, the interaction between PAN and BG sol–gel precursors would influence the micro-structural evolution of BG nanoparticles, while the evolution was definitely related to the silicon content.
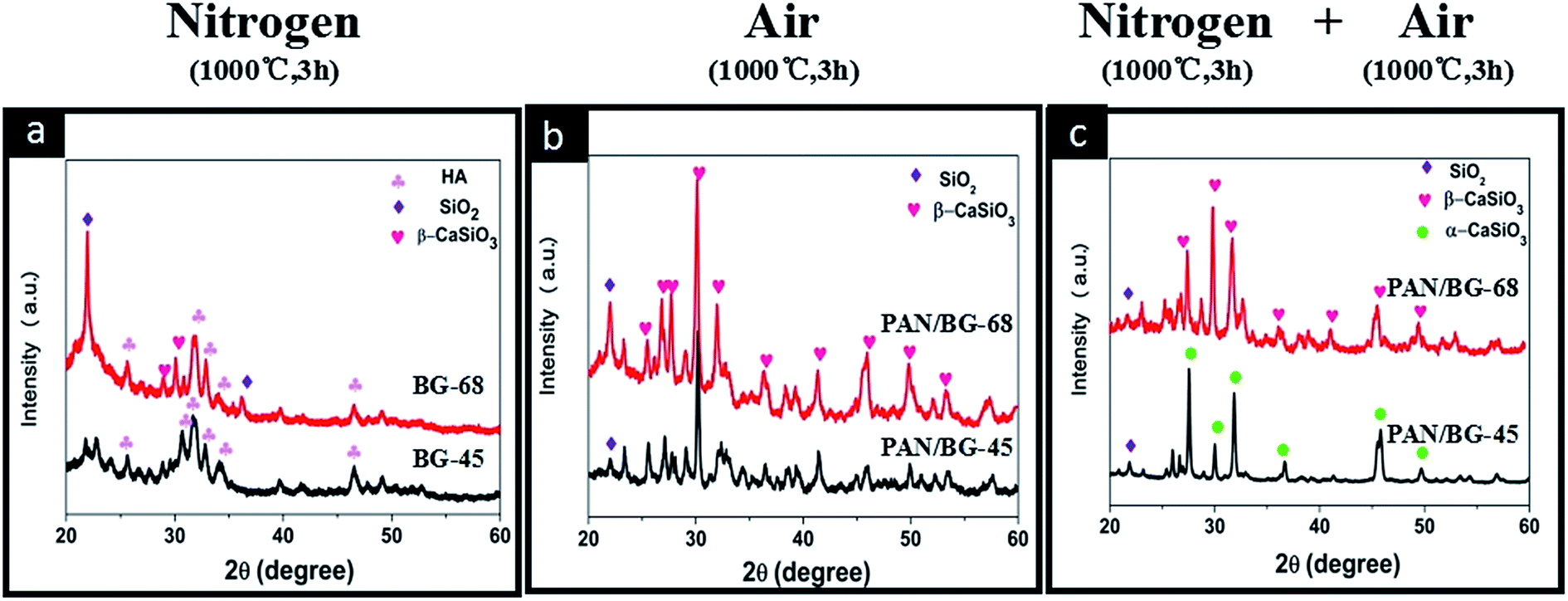 | ||
| Fig. 3 XRD patterns of (a) electrosprayed BG sol–gel sintered in nitrogen, (b) electrospun PAN/BG sol–gel composite nanofibers sintered in air directly and (c) electrospun PAN/BG sol–gel composite nanofibers sintered in nitrogen firstly and then in air. | ||
To confirm the interaction between PAN and BG components, FT-IR and XPS were chosen to characterize the as-electrospun (NF-45 and NF-68), oxidized (ONF-45 and ONF-68) and carbonized (CNF/BG-45 and CNF/BG-68) samples using pure NF, ONF and CNF as reference. As shown in Fig. 4(a1), the peaks at 2243 cm−1 and 1665 cm−1 for as-electrospun NF_were assigned to the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N stretching vibration and the C
N stretching vibration and the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching vibration, respectively.33–35 After BG-45 and BG-68 sol–gel precursor being incorporated, the peak corresponding to CN stretching vibration shifted remarkably from 1665 cm−1 to 1636 cm−1 and 1639 cm−1, respectively. Usually, peak shifting closely related to vibration status, which was influenced by extra interaction. Therefore, the interaction between PAN and BG sol–gel precursor was identified in as-electrospun nanofibers. After the oxidation treatment, in the FT-IR spectrum of ONF (Fig. 4(b1)), a peak at 1592 cm−1 corresponding to the ν ring vibration of N–H in aromatic rings appeared due to the structure transformation of NF deriving from dehydrogenation and cyclization reactions. In the cases of ONF-45 and ONF-68, peak of ν(N–H) was detected only slightly shifting to 1589 cm−1 and 1593 cm−1, respectively, indicating the interaction between oxidized PAN and BG component being weak. After the carbonization treatment, the absorption peak at 1115 cm−1 in CNF was ascribed to the aromatic ring in-plane bending of CC, which illustrated the formation of graphite structure (Fig. 4(c1)). While in the FT-IR spectra of CNF/BG-45 and CNG/BG-68, an obvious absorption peak at 1073 or 1096 cm−1 was detected, indicating the formation of Si–O–Si groups. The difference in the wavenumbers demonstrated subtle difference in the micro-structure of BG component in the two cases. Referring to literature data,36,37 the 1073 cm−1 in CNF/BG-45 was likely ascribed to α-CaSiO3, while the 1096 cm−1 in CNF/BG-68 was likely assigned to β-CaSiO3. In comparing the three FT-IR spectra of electrospun products after different treatments, it was inferred that the interaction between the PAN and the BG sol–gel precursor was most significant during the solution and electrospinning stage as the extent of wavenumber shift shown. XPS results were shown in Fig. 4(a2–c2) to further identify this inference. In comparison with the N1s signal (398.49 eV) in as-electrospun NF, the N1s signal in as-electrospun NF-45 or NF-68 had negatively shifted to 398.17 eV or positively shifted to 399.20 eV (Fig. 4(a2)) when BG sol–gel precursor of different silicon content was incorporated. The results confirmed that the N-containing group in PAN surely had significant interaction with BG component. Also in Fig. 4(a2), the presented Ca2p and Si2p signals in NF-45 and NF-68 gave out different information. It could be seen that the Si2p signals in both cases were almost identifiable at the site of 102.9 eV, while the Ca2p signals were found at 348.19 eV in NF-45 and 347.69 eV in NF-68. This suggested that the electron density around Ca element was different in the two cases, which could be the result of the aforementioned interaction between PAN and BG component, as well as the different silicate networks in NF-45 and NF-68 due to their different silicon content. At the stage of oxidization, the peak shifting in N1s signal was insignificant among samples of ONF, ONF-45 and ONF-68, showing the interaction between oxidized PAN and BG component being weak (Fig. 4(b2)). After carbonization, N1s signal was still detectable in all the three samples although CNFs had formed (Fig. 4(c2)). The reason could be the carbonization temperature (1000 °C) not high enough or carbonization time not long enough to remove all the N element. The insignificant difference in N1s signal among samples of CNF, CNF/BG-45 and CNF/BG-68, however, suggested that no obvious interaction between CNF and BG component existed at this stage. Then, from all these results of Fig. 2 and 4, it was concluded that PAN could interact with BG sol–gel precursor in their mixing and electrospinning process, and the interaction was influenced by the silicon content in the BG sol–gel precursor. Accordingly, different crystalline structures of BG components in the final CNF/BG hybrids developed.
N stretching vibration, respectively.33–35 After BG-45 and BG-68 sol–gel precursor being incorporated, the peak corresponding to CN stretching vibration shifted remarkably from 1665 cm−1 to 1636 cm−1 and 1639 cm−1, respectively. Usually, peak shifting closely related to vibration status, which was influenced by extra interaction. Therefore, the interaction between PAN and BG sol–gel precursor was identified in as-electrospun nanofibers. After the oxidation treatment, in the FT-IR spectrum of ONF (Fig. 4(b1)), a peak at 1592 cm−1 corresponding to the ν ring vibration of N–H in aromatic rings appeared due to the structure transformation of NF deriving from dehydrogenation and cyclization reactions. In the cases of ONF-45 and ONF-68, peak of ν(N–H) was detected only slightly shifting to 1589 cm−1 and 1593 cm−1, respectively, indicating the interaction between oxidized PAN and BG component being weak. After the carbonization treatment, the absorption peak at 1115 cm−1 in CNF was ascribed to the aromatic ring in-plane bending of CC, which illustrated the formation of graphite structure (Fig. 4(c1)). While in the FT-IR spectra of CNF/BG-45 and CNG/BG-68, an obvious absorption peak at 1073 or 1096 cm−1 was detected, indicating the formation of Si–O–Si groups. The difference in the wavenumbers demonstrated subtle difference in the micro-structure of BG component in the two cases. Referring to literature data,36,37 the 1073 cm−1 in CNF/BG-45 was likely ascribed to α-CaSiO3, while the 1096 cm−1 in CNF/BG-68 was likely assigned to β-CaSiO3. In comparing the three FT-IR spectra of electrospun products after different treatments, it was inferred that the interaction between the PAN and the BG sol–gel precursor was most significant during the solution and electrospinning stage as the extent of wavenumber shift shown. XPS results were shown in Fig. 4(a2–c2) to further identify this inference. In comparison with the N1s signal (398.49 eV) in as-electrospun NF, the N1s signal in as-electrospun NF-45 or NF-68 had negatively shifted to 398.17 eV or positively shifted to 399.20 eV (Fig. 4(a2)) when BG sol–gel precursor of different silicon content was incorporated. The results confirmed that the N-containing group in PAN surely had significant interaction with BG component. Also in Fig. 4(a2), the presented Ca2p and Si2p signals in NF-45 and NF-68 gave out different information. It could be seen that the Si2p signals in both cases were almost identifiable at the site of 102.9 eV, while the Ca2p signals were found at 348.19 eV in NF-45 and 347.69 eV in NF-68. This suggested that the electron density around Ca element was different in the two cases, which could be the result of the aforementioned interaction between PAN and BG component, as well as the different silicate networks in NF-45 and NF-68 due to their different silicon content. At the stage of oxidization, the peak shifting in N1s signal was insignificant among samples of ONF, ONF-45 and ONF-68, showing the interaction between oxidized PAN and BG component being weak (Fig. 4(b2)). After carbonization, N1s signal was still detectable in all the three samples although CNFs had formed (Fig. 4(c2)). The reason could be the carbonization temperature (1000 °C) not high enough or carbonization time not long enough to remove all the N element. The insignificant difference in N1s signal among samples of CNF, CNF/BG-45 and CNF/BG-68, however, suggested that no obvious interaction between CNF and BG component existed at this stage. Then, from all these results of Fig. 2 and 4, it was concluded that PAN could interact with BG sol–gel precursor in their mixing and electrospinning process, and the interaction was influenced by the silicon content in the BG sol–gel precursor. Accordingly, different crystalline structures of BG components in the final CNF/BG hybrids developed.
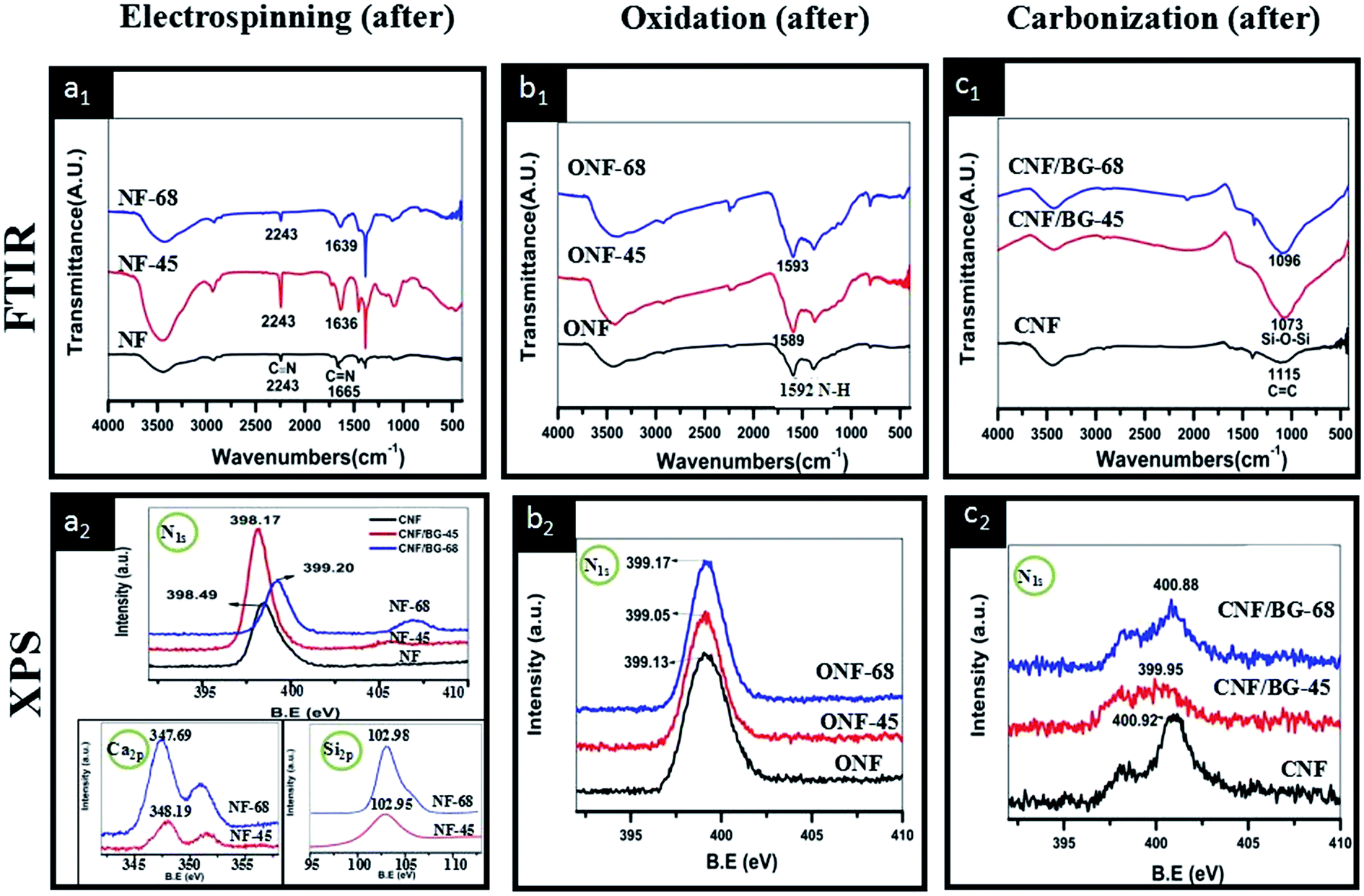 | ||
| Fig. 4 (a1–c1) FTIR and (a2–c2) N1s XPS spectra of CNF and CNF/BG hybrids with different silicon contents at different steps of fabrication process. The lower insets in (a2) are the corresponding Ca2p and Si2p XPS spectra of as-electrospun NF-45 and NF-68. | ||
The zeta potential of pure CNF, CNF/BG-45 and CNF/BG-68 were determined in ethanol instead of physiological saline for avoiding the dissolution of BG component in aqueous solution. As shown in Fig. 5, the zeta potentials of these three samples were all positive, while with different values. Some reports revealed that bioceramics could have positive zeta potential values38 and this feature could influence the attachment or proliferation of bone cells.39,40 Thus, the positive zeta potentials measured for pure CNF and CNF/BG hybrids might play important role in regulating biological properties of bone-related cells. In comparison with the zeta potential of pure CNF, the higher value of CNF/BG-45 and the lower value of CNF/BG-68 indicated that the CNF/BG hybrids had different surface properties when the silicon contents were different. It was suggested that the different composition and crystalline structure might be responsible for their surface features, which would affect the following biomineralization and BMSC biological behaviors in different ways.
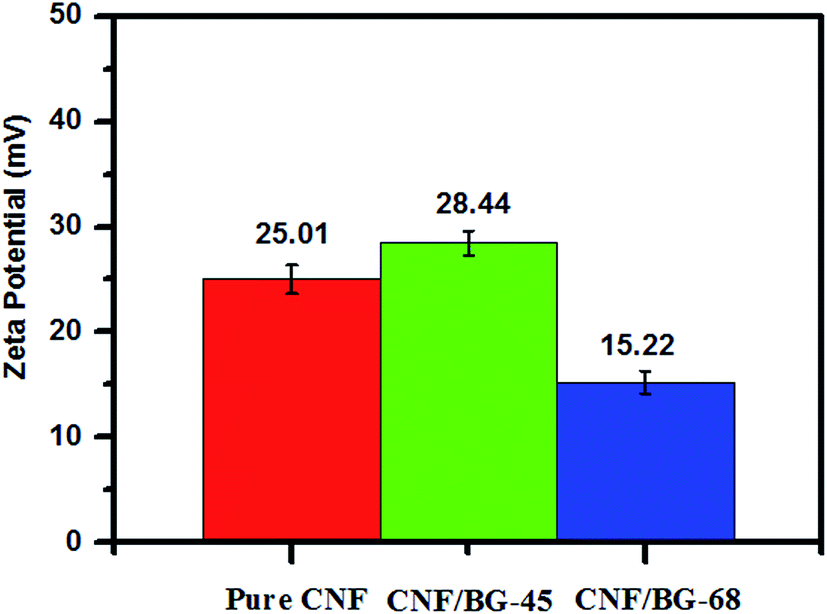 | ||
| Fig. 5 The zeta potential of pure CNF, CNF/BG-45 and CNF/BG-68. | ||
The bioactivity of hybridized CNF/BG depended on the dissolution of BG component, which was influenced by the crystalline structure and the chemical composition of BG nanoparticles, as well as the morphology of CNF/BG.9,10 Since the obtained CNF/BG-45 and CNF/BG-68 had different crystalline structure and chemical composition, their bioactivities should present difference accordingly. The in vitro biomineralization in SBF was applied for the evaluation, in which, the formation of bone-like apatite layer on CNF/BG-45 and CNF/BG-68 was examined using pure CNF as reference. As shown in Fig. 6, CNF/BG hybrids demonstrated much stronger ability in inducing mineral deposition than pure CNFs after being soaked in SBF for 3–7 days. Only scattered mineral aggregates were observed on pure CNFs at the seventh day of biomineralization, while both CNF/BG hybrids had been fully covered by mineral depositions at the same time point. Clearly, it was the incorporation of BG component having endowed the CNFs material excellent bioactivity. While different mineral deposition rate was also able to be observed between CNF/BG-45 and CNF/BG-68. Comparing Fig. 6(b2 and c2) or (b3 and c3), the amount of mineral deposition on CNF/BG-45 was apparently higher than that on CNF/BG-68. It was suggested that the dissolution rate of α-CaSiO3 in CNF/BG-45 was faster than that of β-CaSiO3 in CNF/BG-68. As shown in Fig. 6(b1 and c1), those BG nanoparticles on fiber surface disappeared within 1 day of SBF soaking due to their dissolution in aqueous solution. The ion release from CNF/BG hybrids provided nucleation sites for new mineral growth. From Fig. 2(a1 and b1), the BG nanoparticles was smaller in size in CNF/BG-45 than in CNF/BG-68. This could be one reason to cause faster dissolution of BG component in CNF/BG-45 than in CNF/BG-68. On the other hand, the structure of α-CaSiO3 in CNF/BG was reported pseudo-hexagonal consisted of three covalently bonded silicate tetrahedra via corner-sharing oxygens to form “3-rings” structure, while β-CaSiO3 was composed of corner-sharing silicate tetrahedral linked structure to form chains.41,42 The high strain associated with the Si–O–Si bond angles in the “3-rings” of α-CaSiO3 made these bonds more susceptible to hydrolysis compared to the chain-like Si–O–Si bonds in β-CaSiO3. Therefore, CNF/BG-45 induced faster mineral nucleation and growth than CNF/BG-68. The deposited minerals were characterized with FT-IR and XRD. In the FT-IR spectra (insets in Fig. 6(a2–c2)), characteristic signals (585, 606 and 1380 cm−1) assigned to phosphate groups could be identified.43 In the XRD spectra (insets in Fig. 6(a3–c3)), characteristic diffraction peaks (2θ = 25° and 32°) assigned to (0 0 2) and (2 1 1) reflections of crystalline HA, respectively, were detected.44 The FT-IR and XRD results revealed that the mineral depositions were mainly HA, which was the dominant component in natural bone minerals. At the seventh day of biomineralization, the mineral on CNF/BG had presented a kind of characteristic flaky-like structure of HA.
 | ||
| Fig. 6 SEM images of (a) pure CNF, (b) CNF/BG-45 and (c) CNF/BG-68 hybrids being soaked in SBF for 1, 3, 5 and 7 days at 37 °C. The insets are the FTIR spectra (second column), XRD patterns (third column) and high-magnification SEM images (fourth column) obtained from mineral depositions on corresponding samples at corresponding time points. | ||
Subsequently, BMSCs were cultured on pure CNF and CNF/BG hybrids to evaluate the effect of BG components on cell proliferation and osteogenic differentiation. As shown in Fig. 7(a), continuous cell proliferation was detected on all the CNF-based materials, indicating the biocompatibility of the materials. However, the growth of BMSCs could be seen significantly faster on both the CNF/BG-45 and CNF/BG-68 than on the pure CNF. Besides, CNF/BG-45 could enhance the cell proliferation more efficiently than CNF/BG-68. These results were consistent with the biomineralization trend in SBF. It was proposed that CNF/BG hybrids were more bioactive than pure CNF owing to the incorporation of BG component. Moreover, CNF/BG-45 dissolved faster than CNF/BG-68, which was able to provide more bioactive ions like Ca2+ to promote cell proliferation.45,46 As for the ALP activity, the expression reached the maximum value at day 14 on all the materials (Fig. 7(b)). Undoubtedly, the two CNF/BG hybrids had significantly enhanced the ALP activity of BMSCs in comparison with pure CNF, because they could provide bioactive components to favor the osteogenic differentiation of BMSCs via the dissolution of incorporated BG nanoparticles.47,48 In a whole, the CNF/BG-68 could promote the expression of ALP activity slightly higher than CNF/BG-45, while the difference in between them was insignificant. As shown in Fig. 6(b4 and c4), it could be seen the apatite deposition was also similar on both the CNF/BG-45 and CNF/BG-68 after 7 days biomineralization. The reason for this phenomenon was ascribed to the less difference in the contents of soluble ingredients released from the CNF/BG hybrids after the initial fast dissolution.
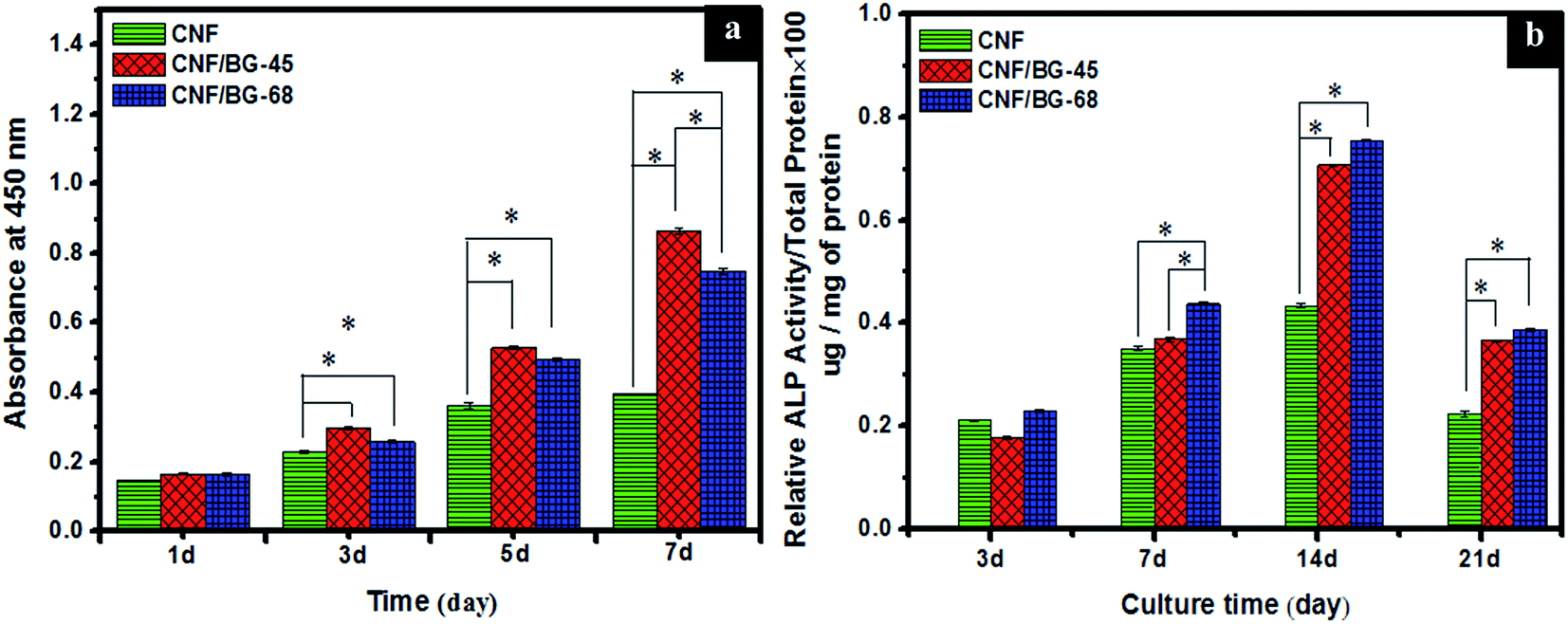 | ||
| Fig. 7 (a) Cell proliferation analyzed using CCK-8 method by the absorbance measured at 450 nm for the indicated samples for 1–7 days and (b) ALP activity expression of BMSCs cultured on pure CNF, CNF/BG-45 and CNF/BG-68 for various times. The results are represented as mean ± standard deviation for n = 3 (P < 0.05). *P < 0.05, significantly different. | ||
In summary, the silicon content in BG sol–gel precursor could be a preparation parameter in regulating the bioactivity of CNF/BG hybrids. Due to the interaction between PAN component and BG component in solution electrospinning, BG nanoparticles with different crystalline structures were resulted from their different initial feeding dose of BG precursors. Thus, the dissolution ability or rate of BG nanoparticles in final CNF/BG hybrids was different, which led to different ability in inducing apatite formation and in enhancing osteocompatibility to obtain promising substrates for bone repairing.
Acknowledgements
The authors are very pleased to acknowledge financial support from National Basic Research Program of China (2012CB933904), National Natural Science Foundation of China (51373016 and 51473016), the Research Fund for the Doctoral Program of Higher Education (No. 20110010120014), and the Fundamental Research Funds for the Central Universities (No. ZY1106).Notes and references
- L. L. Tan, X. M. Yu, P. Wan and K. Yang, J. Mater. Sci. Technol., 2013, 29, 503 CAS.
- Y. C. Liu, J. Lim and S. H. Teoh, Biotechnol. Adv., 2013, 31, 688 CrossRef CAS PubMed.
- N. R. Mohamed, E. D. Delbert, B. B. Sonny, F. Qiang, B. J. Steven, F. B. Lynda and P. T. Antoni, Acta Biomater., 2011, 7, 2355 CrossRef PubMed.
- K. A. Hing, P. A. Revell, N. Smith and T. Buckland, Biomaterials, 2006, 27, 5014 CrossRef CAS PubMed.
- J. R. Jones, Acta Biomater., 2013, 9, 4457 CrossRef CAS PubMed.
- I. Rajzer, E. Menaszek, L. Bacakova, M. Rom and M. Blazewicz, J. Mater. Sci.: Mater. Med., 2010, 21, 2611 CrossRef CAS PubMed.
- A. Fraczek-Szczypta, S. Rabiej, G. Szparaga, E. Pabjanczyk-Wlazlo, P. Krol, M. Brzezinska, S. Blazewicz and M. Bogun, Mater. Sci. Eng., C, 2015, 51, 336 CrossRef CAS PubMed.
- I. Rajzer, R. Kwiatkowski, W. Piekarczyk, W. Biniaś and J. Janicki, Mater. Sci. Eng., C, 2012, 32, 2562 CrossRef CAS.
- X. L. Jia, T. H. Tang, D. Cheng, C. H. Zhang, R. Zhang, Q. Cai and X. P. Yang, Colloids Surf., B, 2015, 136, 585 CrossRef CAS PubMed.
- C. H. Zhang, D. Cheng, T. H. Tang, X. L. Jia, Q. Cai and X. P. Yang, J. Mater. Chem. B., 2015, 3, 5300 RSC.
- X. L. Jia, T. H. Tang, D. Cheng, L. J. Guo, C. H. Zhang, Q. Cai and X. P. Yang, RSC Adv., 2014, 4, 64299 RSC.
- K. A. Hing, P. A. Revell, N. Smith and T. Buckland, Biomaterials, 2006, 27, 5014 CrossRef CAS PubMed.
- T. Kokubo and H. Takadama, Biomaterials, 2006, 27, 2907 CrossRef CAS PubMed.
- K. Lin, J. Chang, X. G. Liu, L. Chen and Y. L. Zhou, CrystEngComm, 2011, 13, 4850 RSC.
- C. S. Johnson and D. A. Gabriel, Laser light scattering, Dover publications, INC., New York, 1994 Search PubMed.
- M. Elimelech, J. Gregory, X. D. Jia and R. Williams, Particle deposition and aggregation: Measurement, Modelling and Simulation, Butterworth-Heinemann, Woburn, 1998 Search PubMed.
- X. L. Jia, J. Listak, W. Velencia, E. E. Kalu, X. P. Yang and M. Bockstaller, Langmuir, 2010, 26, 12190 CrossRef CAS PubMed.
- C. Vaid, S. Murugavel, C. Das and S. Asokan, Microporous Mesoporous Mater., 2014, 186, 46 CrossRef CAS.
- F. He, H. L. Zhao, X. H. Qu, C. J. Zhang and W. H. Qiu, J. Mater. Process. Technol., 2009, 209, 1621 CrossRef CAS.
- G. Poologasundarampillai, B. Yu, J. R. Jones and T. Kasug, Soft Matter, 2011, 7, 10241 RSC.
- W. J. Gao, Y. Wan, Y. Q. Dou and D. Y. Zhao, Adv. Energy Mater., 2011, 1, 115 CrossRef CAS.
- Z. P. Zhou, K. M. Liu, C. L. Lai, L. F. Zhang, J. H. Li, H. Q. Hou, D. H. Reneker and H. Fong, Polymer, 2010, 51, 2360 CrossRef CAS.
- H. Wang, S. C. Zhao, J. Zhou, Y. Q. Shen, W. H. Huang, C. Q. Zhang, M. N. Rahamanc and D. P. Wang, J. Mater. Chem. B, 2014, 2, 8547 RSC.
- F. Pishbin, V. Mouriño, S. Flor, S. K. V. Salih, M. P. Ryan and A. R. Boccaccini, ACS Appl. Mater. Interfaces, 2014, 6, 8796 CAS.
- X. Zhang, X. W. Li, J. G. Li and X. D. Sun, ACS Appl. Mater. Interfaces, 2014, 6, 513 CAS.
- P. Siriphannon, Y. Kameshima, A. Yasumori, K. Okada and S. Hayashi, J. Eur. Ceram. Soc., 2002, 22, 511 CrossRef CAS.
- L. M. Rodríguez-Lorenzo and M. Vallet-Regı, Chem. Mater., 2000, 12, 2460 CrossRef.
- T. Sopcak, L. M. V. Girman and J. Durisin, J. Non-Cryst. Solids, 2015, 415, 16 CrossRef CAS.
- F. S. Shirazi, M. Mehrali, A. A. Oshkour, H. S. C. Metselaar, N. A. Kadri and N. A. AbuOsman, J. Mech. Behav. Biomed. Mater., 2014, 30, 168 CrossRef CAS PubMed.
- H. C. Li, D. G. Wang, C. Z. Chen and H. Shi, Mater. Lett., 2015, 159, 459 CrossRef CAS.
- H. H. Beheri, K. R. Mohamed and G. T. El-Bassyouni, Mater. Des., 2013, 44, 461 CrossRef CAS.
- K. Lin, D. Zhai, N. Zhang, N. Kawazoe, G. P. Chen and J. Chang, Ceram. Int., 2014, 40, 3287 CrossRef CAS.
- F. Li, W. M. Kang, B. Cheng and Y. C. Dong, Catal. Commun., 2015, 69, 150 CrossRef CAS.
- W. X. Zhang, J. Liu and G. Wu, Carbon, 2003, 41, 2805 CrossRef CAS.
- M. Y. Wu, Q. Y. Wang, K. Li, Y. Q. Wu and H. Q. Liu, Polym. Degrad. Stab., 2012, 97, 1511 CrossRef CAS.
- M. Catauro, F. Bollino, R. A. Renella and F. Papale, Ceram. Int., 2015, 41, 12578 CrossRef CAS.
- W. Holand, W. Vogel, K. Naumann and J. Gummel, J. Biomed. Mater. Res., 1985, 19, 303 CrossRef CAS PubMed.
- K. Cheng, W. Weng, H. Wang and S. Zhang, Biomaterials, 2005, 26, 6288 CrossRef CAS PubMed.
- A. Fahami, G. W. Beall and T. Betancourt, Mater. Sci. Eng., C, 2016, 59, 78 CrossRef CAS PubMed.
- R. Smeets, A. Kolk, M. Gerressen, O. Driemel, O. Maciejewski, B. Hermanns-Sachweh, D. Riediger and J. M. Stein, Head Face Med., 2009, 5, 13 Search PubMed.
- X. Zhang, X. W. Li, J. G. Li and X. D. Sun, ACS Appl. Mater. Interfaces, 2014, 6, 513 CAS.
- W. Xia and J. Chang, Microporous Mesoporous Mater., 2008, 18, 345 CrossRef.
- J. L. Arias, F. J. García-Sanz, M. B. Mayor, S. Chiussi, J. Pou, B. León and M. Pérez-Amor, Biomaterials, 1998, 19, 883 CrossRef CAS PubMed.
- R. Govindan, G. S. Kumarand and E. K. Girija, RSC Adv., 2015, 5, 60188 RSC.
- A. A. R. De Oliveira, D. A. De Souza, L. L. S. Dias, S. M. de Carvalho, H. S. Mansur and M. P. Marivalda, Biomed. Mater., 2013, 8, 025011 CrossRef PubMed.
- A. Hoppe, N. S. Guldal and A. R. Boccaccini, Biomaterials, 2011, 32, 2757 CrossRef CAS PubMed.
- V. V. Valimaki, J. J. Yrjans, E. L. Vuorio and H. T. Aro, Tissue Eng., 2005, 11, 387 CrossRef PubMed.
- L. L. Hench, J. Eur. Ceram. Soc., 2009, 29, 1257 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |