
Thiophene-S,S-dioxidized indophenine (IDTO) based donor–acceptor polymers for n-channel organic thin film transistors†
Abstract
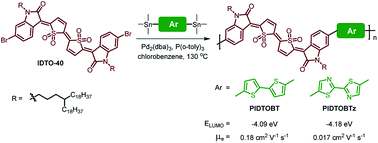
Two donor–acceptor (D–A) conjugated polymers, PIDTOBT and PIDTOBTz, based on thiophene-S,S-dioxidized indophenine (IDTO) as the acceptor building block are synthesized for solution processed organic thin-film transistors (OTFTs). The influences of the donor unit on the photophysical, electrochemical and electron-transport properties were investigated. These polymers possess very deep highest-occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energy levels due to the strong electron accepting capability of the IDTO moiety. In OTFT devices, both polymers exhibited unipolar n-type charge transport characteristics with electron mobility up to 0.18 cm2 V−1 s−1.
 Please wait while we load your content...
Please wait while we load your content...

