
Controlled synthesis of concentration gradient LiNi0.84Co0.10Mn0.04Al0.02O1.90F0.10 with improved electrochemical properties in Li-ion batteries†
Weihua Chen*a,
Yanyang Lia,
Juanjuan Zhaoa,
Feifei Yanga,
Jianmin Zhanga,
Qiuzhi Shia and
Liwei Mi*b
aCollege of Chemistry and Molecular Engineering, Zhengzhou University, Zhengzhou, 450001, P. R. China. E-mail: chenweih@zzu.edu.cn
bCenter for Advanced Materials Research, Zhongyuan University of Technology, Zhengzhou, 450007, P. R. China. E-mail: mlwzzu@163.com
First published on 2nd June 2016
Abstract
For Ni-based core–shell materials, a separation may occur between the coating layers and bulk material in the charge–discharge process because of the phase difference. Therefore, concentration gradient materials have higher cycling stability than core–shell materials. In this study, a concentration gradient material, LiNi0.84Co0.10Mn0.04Al0.02O1.90F0.10, was achieved by an ion exchange method. In the synthesized LiNi0.84Co0.10Mn0.04Al0.02O1.90F0.10, the distribution of the metal elements Al, Ni and Mn is a radial gradient, as confirmed by line scanning on the cross section of a single particle of the sample, whereas Co, O and F show almost uniform distribution. As a cathode material of Li-ion batteries, the capacity retention after 40 cycles of the synthesized LiNi0.84Co0.10Mn0.04Al0.02O1.90F0.10 with low cost is 1.38 times that of LiNi0.83Co0.10Mn0.07O2. Furthermore, the differential scanning calorimetry result shows that LiNi0.84Co0.10Mn0.04Al0.02O1.90F0.10 demonstrates a much lower amount of exothermic heat release than LiNi0.83Co0.10Mn0.07O2. In addition, the cycling stability of LiNi0.84Co0.10Mn0.04Al0.02O1.90F0.10 is better than those of LiNi0.83Co0.10Mn0.05Al0.02O2 and LiNi0.85Co0.10Mn0.05O1.97F0.03 prepared via the same method. All of the abovementioned materials are calcined in air with low cost and show somewhat low discharge capacity. However, for the materials obtained from calcining Ni0.84Co0.10Mn0.04Al0.02(OH)1.9F0.10 in O2 with higher cost, the discharge capacity increases from 140.3 mA h g−1 for LiNi0.84Co0.10Mn0.04Al0.02O1.90F0.10 calcined in air to 177.5 mA h g−1. To the best of our knowledge, this is the first time that cation Al3+ and anion F− have been introduced via an ion exchange method to improve the electrochemical properties of high Ni content cathode materials. This study provides a simple way to synthesize hierarchical micro-spherical high Ni content cathode with a gradient distribution of elements in air.
Introduction
Li-ion batteries have been widely used in portable electronic devices and electric vehicles.1–3 During their applications, the cycle life of batteries is one of the most important aspects to be considered. To improve the cycle stability of electrode materials, many strategies have been extensively investigated.4–17 As advanced cathode materials for Li-ion batteries, LiNiO2-based cathode materials have drawn significant attention because of their high energy density and relatively low cost. However, their insufficient cycle life and poor thermal stability prevent their practical applications.3 Also, materials with high Ni content (>0.7) are difficult to achieve at low oxygen partial pressures.18 In general, a coating approach can stabilize the electrode–electrolyte interfaces markedly. For example, a thin insulating AlF3 coating can protect active materials from electrolyte damage under high voltage or large current and suppress exothermic reactions with the liquid electrolyte.19,20 The modification of active cathode materials by doping is greatly effective with respect to structural stability in a highly oxidized state. The doping of Al3+ and F− plays a supporting role in the structure and enhances the electrochemical properties.21,22 However, both of these methods have their own weaknesses.23 For the coating, separation may occur between the coating layers and bulk material because of the phase difference, which is detrimental to the cycle performance.24,25 Similarly, in doped materials, the capacities are limited by the electrochemically inactive element. In LiNiO2-based cathode materials, the high Ni content would provide high capacity along with serious capacity fading. Contrastingly, materials with high content of non-electrochemically active elements (Mn, Al) have good cycle performance and thermostability; however, they have low discharge capacity. Homogeneous Ni–Co–Mn and Ni–Co–Al multivariant transition metal oxide cathode materials usually cannot meet the needs of both aspects. However, concentration gradient materials can address this problem.Materials with concentration gradient architectures were designed to enhance the cycle life of Li-ion batteries by Sun et al.24,26 Within each particle of the concentration gradient material, the nickel-enriched inner core delivers high capacity, and the manganese-rich shell provides outstanding cycle life and safety. Since then, a series of concentration gradient materials have been synthesized by coprecipitation.27,28 Compared with conventional coating and doping, the method of structuring the concentration gradient can provide high stability and good properties. Nevertheless, the approach of coprecipitation has great limitations itself, because it requires similar solubility of all the compounds and cannot be applied to establish the anion gradient.
Cation exchange is a simple diffusion process initially applied to synthesize a broad range of inorganic nanocrystals.29 This process can be used to achieve rapid and low-temperature transformations in the solid state.30,31 A core–shell Ni(OH)2@CoOOH composite has been constructed via a simple cation exchange route under moderate conditions.32 In our previous reports,22 cation exchange was also adopted to promote the discharge capacities of the cathode material LiNi0.83Co0.10Mn0.07O2. This reaction has been further applied to anions. Saruyama et al.33 synthesized anisotropically phase-segregated CdS/CdTe heterodimers via an anion exchange reaction of CdS NPs with an organic telluride. Using specific reaction conditions, ion exchange can be applied to synthesize unique nanocrystals and core–shell particles. Therefore, ion exchange can be used to construct concentration gradient materials and introduce aluminum and fluorine ions into the materials, which can stabilize the crystal structure and promote resistance to corrosion from the environment.34–36
Here, the anion and cation exchange method was adopted to design microsphere materials with a concentration gradient of metals. The ion exchange occurred between Ni0.83Co0.1Mn0.07(OH)2 and Al3+, F− under hydrothermal conditions. Then, the concentration gradient hydroxides Ni0.84Co0.10Mn0.04Al0.02(OH)1.9F0.10 and LiOH·H2O were mixed and calcined to synthesize the corresponding concentration gradient material LiNi0.84Co0.10Mn0.04Al0.02O1.90F0.10. The process of calcination was completed in air to explore a simplified method for high Ni content materials. As the cathode of Li-ion batteries, the designed hierarchical micro-spherical LiNi0.84Co0.10Mn0.04Al0.02O1.90F0.10 shows improved cycling stability.
Experimental
All of the chemical reagents in this study were of analytical grade and were used without further purification.The spherical precursor Ni0.83Co0.10Mn0.07(OH)2 was prepared by employing a hydrothermal method in which Ni(NO3)2·6H2O, Co(NO3)2·6H2O, 50% Mn(NO3)2, and CON2H4 were used as the starting materials. These materials were dissolved in a mixture of ethanol and distilled water. After the obtained solution was treated by ultrasonication for 0.5 h, it was transferred to a 90 mL Teflon-lined stainless steel autoclave, maintained at 180 °C for 7.5 h, and then cooled to room temperature. The precursor was centrifuged, washed four times with distilled water, and oven-dried at 70 °C overnight. The obtained precursor Ni0.83Co0.1Mn0.07(OH)2 was named MO. A mixture of Ni0.83Co0.1Mn0.07(OH)2 and LiOH·H2O with a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.05 was heated at 500 °C for 5 h and annealed at 650 °C for 12 h in air to prepare Li(Ni0.83Co0.1Mn0.07)O2 powder. The primary active material, Li(Ni0.83Co0.1Mn0.07)O2, was designated as LMOA.
:1.05 was heated at 500 °C for 5 h and annealed at 650 °C for 12 h in air to prepare Li(Ni0.83Co0.1Mn0.07)O2 powder. The primary active material, Li(Ni0.83Co0.1Mn0.07)O2, was designated as LMOA.
AlF3·3H2O, LiF, or Al(NO3)3·9H2O (3 at% of MO) was dissolved in 50 mL distilled water to react with Ni0.83Co0.1Mn0.07(OH)2 for introducing Al3+, and F−, F−, or Al3+. Ni0.83Co0.1Mn0.07(OH)2 (0.25 g) powder was immersed into the solutions of AlF3·3H2O, LiF, or Al(NO3)3·9H2O, respectively, and the mixtures were stirred constantly for 0.5 h at room temperature; afterward, the mixtures were transferred to a 90 mL Teflon-lined stainless steel autoclave and heated at 160 °C for 4 h. After filtration, cleanout and desiccation was performed. The obtained Ni0.84Co0.10Mn0.04Al0.02(OH)1.9F0.10, Ni0.85Co0.10Mn0.05(OH)1.97F0.03 and Ni0.83Co0.10Mn0.05Al0.02(OH)2 were named AFMO, FMO and AMO, respectively. The obtained hydroxides were calcined with LiOH·H2O under air atmosphere at 650 °C. These lithiated oxides, LiNi0.84Co0.10Mn0.04Al0.02O1.90F0.10, LiNi0.85Co0.10Mn0.05O1.97F0.03 and LiNi0.83Co0.10Mn0.05Al0.02O2, obtained in air were named AFLMOA, FLMOA, and ALMOA, respectively. Especially, AFMO and LiOH·H2O were also calcined in O2. The obtained material was named AFLMOO. In addition, ion exchanges with different AlF3 quantities (5 at% and 8 at%) at different temperatures (120, 140, and 180 °C) were explored.
Inductively coupled plasma (ICP; ICAP6500) was used to determine the contents of the metal elements. Powder X-ray diffraction (XRD; Bruker D8 Advance) was employed to characterize the crystal structures of the powder samples by applying Cu-Kα radiation in the 2θ range of 15° to 80° with a continuous scan mode at a scanning rate of 0.1° s−1 at room temperature. The valence state and the chemical environment of the elements on the surface of AFMO were confirmed by X-ray photoelectron spectroscopy (XPS; Kratos AXIS ULTRA). The particle shape and morphological characteristics of the materials were observed by scanning electron microscopy (SEM; ZEISS Merlin Compact). Energy-dispersive X-ray spectroscopy (EDS; Oxford X-Max) was employed to analyze the elemental composition and distribution on the surfaces and cross sections of the micro-spheres. To obtain a cross section of a single micro-sphere, the materials AFMO and AFLMOA were embedded in ethyl α-cyanoacrylate and polished with metallographic sandpapers. Differential scanning calorimetry (DSC; TA MDSC 2920) was performed to compare the thermal stability of LMOA and AFLMOA at the 4.5 V charged state.
The cathodes were prepared by mixing 85.0 wt% active material with 12.0 wt% acetylene black and 3.0 wt% polytetrafluoroethylene. The electrochemical performance of the cathodes was evaluated using coin-type cells, CR2016, in which 1 M LiPF6 dissolved in a 1:1 (v/v) mixture of ethylene carbonate/dimethyl carbonate (EC/DMC) was used as the electrolyte. Celgard 2400 membrane was used as the separator and Li metal foil was used as the counter and reference electrodes. Cycling tests were then performed with a battery test system (LAND CT2001A Model, Wuhan Land Electronics, Ltd.) between 3.0 and 4.5 V at room temperature. Cyclic voltammogram (CV) tests were subsequently performed at a scanning rate of 0.1 mV s−1 between 3.0 and 4.5 V at room temperature on an electrochemical workstation (CHI 604E).
Results and discussion
A program was designed to construct the concentration gradient LiMO2 (M = Ni, Co, and Mn). The schematic of the synthetic process is shown in Scheme 1. As shown in the top left corner of Scheme 1, Al3+ and F− can exchange simultaneously with the cation and anion of the metal hydroxide precursor MO. The reaction is expressed in eqn (1). The driving force of the ion exchange is the difference of the solubility products (Ksp). As shown in Table 1, the solubility product of Al(OH)3 is lower than that of Ni(OH)2, Co(OH)2 or Mn(OH)2. At the beginning of the ion exchange process for AFMO, only Al3+ and F− exist in the neutral solution. Driven by the difference of the solubility products, the Ni2+, Co2+ and Mn2+ in LiNi0.83Co0.10Mn0.07O2 would be exchanged by Al3+ with higher concentration in the solution and transferred into the solution. Similarly, partial OH− could be exchanged by F−. Furthermore, the ion exchange process is also correlated with some factors, such as deepness, contact area, temperature, and concentration of Al3+. Because the outer layer of the micro-spheres has a shorter reaction path and a bigger contact area, the ion exchange would occur more quickly in the outer layer. By controlling the reaction time and temperature of the ion exchange process, the Al3+ would be enriched in the outer layer of the micro-spheres.| M(OH)2 + xAl3+ + yF− ↔ M1−1.5xAlx(OH)2−yFy + 1.5xM2+ + yOH− | (1) |
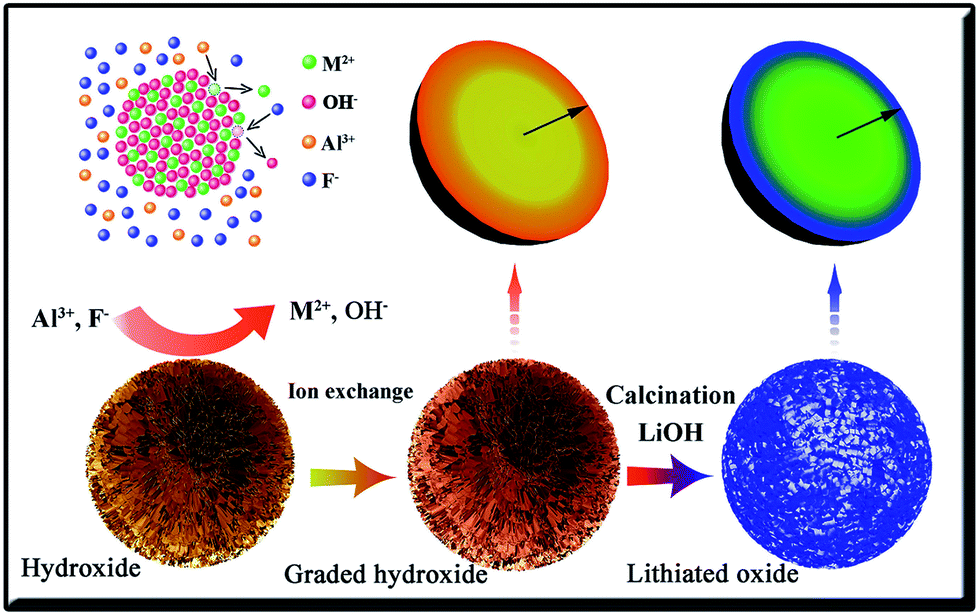 | ||
| Scheme 1 Schematic view of the synthetic process for the concentration gradient materials. | ||
| Compound | Solubility product |
|---|---|
| Ni(OH)2 | 2.0 × 10−15 |
| Co(OH)2 | 1.09 × 10−15 |
| Mn(OH)2 | 1.9 × 10−13 |
| Al(OH)3 | 1.3 × 10−33 |
After ion exchange was performed, the morphology of the micro-spheres shows no evident changes in characteristics. The concentration gradient hydroxide was subsequently calcined with lithium hydroxide to obtain the final concentration gradient lithiated oxide. The final obtained materials also retained the element gradient in each particle. The concentrations of Al and Mn gradually increased from the core to the surface; this finding is in contrast to the distribution of Ni. Meanwhile, the content of Co remained almost constant. The ICP results are shown in Table 2. After ion exchange, the Al3+ is introduced into AFMO successfully. The increased Ni-content in FMO may cause the dissolution of Mn(OH)2. Moreover, the proportion of O and F in AFMO is 1.906/0.094, according to the EDS results.
| Sample | Molar ratio of the sample (%) | |||
|---|---|---|---|---|
| Ni | Co | Mn | Al | |
| MO | 0.828 | 0.101 | 0.071 | 0 |
| AFMO | 0.835 | 0.097 | 0.044 | 0.024 |
| FMO | 0.852 | 0.098 | 0.050 | 0 |
| AMO | 0.834 | 0.097 | 0.047 | 0.022 |
The initial precursor MO, the concentration gradient AFMO, and the lithiated LMOA, AFLMOA, FLMOA and ALMOA were characterized by XRD. The XRD results are shown in Fig. 1. MO and AFMO yield similar diffraction peaks at 2θ of 11.4°, 22.7°, 33.5°, 34.4°, 38.8°, 46.0°, 60.0° and 61.3°, which could be indexed as nickel hydroxide (JCPDS card no. 38-715). This result suggests that the structure of the initial precursor is maintained after ion exchange treatment. The lithiated materials display diffraction peaks at 2θ of 18.8°, 36.6°, 38.1°, 38.3°, 44.4°, 48.9°, 58.8°, 64.470°, 64.7°, 68.0°, 76.7° and 77.5°. All of the peaks could be identified, revealing characteristics similar to the hexagonal α-NaFeO2 structure (JCPDS card no. 09-0063). Compared with those of LMOA, the (104) peaks of AFLMOA and FLMOA shift to a low degree, which indicates the substitution element F has been doped into the crystal structure.37,38 The lattice parameters of the four samples have been calculated by Jade 6, as shown in Table 3. The lattice parameters of AFLMOA, ALMOA and FLMOA are bigger than those of LMOA. This result indicates that Al3+ and F− have been introduced into the crystal lattice.21 The mean crystallite sizes of the four samples have been calculated by Jade 6, as shown in Table 4. The mean crystallite sizes of FLMOA and ALMOA are smaller than that of LMOA. AFLMOA has the smallest mean crystallite size among the four samples. The introduction of Al3+ and F− could reduce the mean crystallite sizes of the samples.
 | ||
| Fig. 1 The XRD patterns of MO, AFMO, LMOA, AFLMOA, FLMOA and ALMOA. Partial enlarged view of the 104 peaks of LMOA, AFLMOA, FLMOA and ALMOA. | ||
| Materials | a/Å | c/Å | V/Å3 |
|---|---|---|---|
| LMOA | 2.876 | 14.178 | 101.56 |
| AFLMOA | 2.880 | 14.188 | 101.89 |
| FLMOA | 2.880 | 14.183 | 101.89 |
| ALMOA | 2.876 | 14.181 | 101.60 |
| Sample | Mean crystallite size (nm) |
|---|---|
| LMOA | 19.1 |
| AFLMOA | 16.4 |
| FLMOA | 17.7 |
| ALMOA | 17.5 |
The morphological characteristics of MO, AFMO, and AFLMOA were observed by SEM [Fig. 2(a–c)]. MO and AFMO (approximately 10 μm in size) consist of primary sheet particles, which confirms their hierarchical micro-spherical structure. As in a previous report,22 the morphological characteristics of these particles are inherited after ion exchange, which is an advantage of ion exchange compared with other methods. The corresponding material AFLMOA maintains the spherical morphology, with a 3D hierarchical microstructure. The hierarchical microstructure is composed of thick plates, which are transformed from hydroxide nanoflakes. The EDS result of the AFLMOA powder was recorded in Fig. 2(d). The existence of peaks corresponding to Al and F indicates that Al3+ and F− are successfully introduced into the metal hydroxide. The elemental mapping of AFLMOA was obtained using SEM-EDS. The differently colored spots represent different elements, and the spot intensity is an indicator of the element concentration. Indeed, the Ni, Co, Mn, Al, and F contents are uniformly distributed on the surface. Additional SEM and EDS results for MO, FMO, and AMO are shown in Fig. S1 and S2.†
 | ||
| Fig. 2 SEM images of MO (a), AFMO (b), AFLMOA (c), the EDS results (d) and the element mapping on the surface of a single micro-sphere of AFLMOA. | ||
The surface of AFMO was tested by XPS to investigate the chemical environment of each element and elucidate the reaction mechanism of ion exchange. The observed (black scatter plot) pattern and the calculated (red line) pattern are well matched, indicating that the fitting results are reliable. The peaks assigned to Ni 2p3/2, Ni 2p1/2, Co 2p3/2, Co 2p1/2, Mn 2p3/2, Mn 2p1/2, Al 2p and F 1s are identified in the survey spectra.
The peaks at 856.1 and 873.7 eV are assigned to Ni2+.39 The peaks at 781.4 and 797.0 eV are assigned to Co2+.40 The peaks at 641.3 and 652.8 eV are assigned to Mn3+.41 The appearance of Mn3+ on the surface should be caused by the oxidation of air. Fortunately, the valence state of the Mn ion is unlikely to impede subsequent synthesis. The final lithiated material can also be synthesized from Mn3+. In Fig. 3(e), the peak at 68.0 eV is assigned to Ni 3p.42 The peak at 70.6 eV should be thought as a ghost peak. The peak at 74.1 eV is assigned to Al 2p, which is close to the binding energy of Al 2p in Al(OH)3 (74.2 eV) and far from the binding energy of Al 2p in AlF3 (76.9 eV).43 This result indicates that Al is not directly bound to F in AFMO, but is bound to OH−. Meanwhile, the binding energy of F 1s in AFMO is 684.2 eV, which is close to the binding energy of F 1s in NiF2 (684.5 eV)44 and obviously different from the binding energy of F 1s in AlF3 (687.8 eV).44 The result further confirms the lack of binding between F and Al in AFMO. Based on this analysis, the XPS results of AFMO confirm that Al3+ and F− have been introduced simultaneously to the precursor MO via exchange with the cations (metal ions) and anion, respectively.
 | ||
| Fig. 3 XPS spectra and the corresponding fitting curves of AFMO. Survey spectra (a), Ni 2p spectra (b), Co 2p spectra (c), Mn 2p spectra (d), Al 2p spectra (e) and F 1s spectra (f). | ||
To confirm the distribution of the elements in the interior of the micro-spheres, AFMO and AFLMOA were embedded in ethyl α-cyanoacrylate and polished with metallographic sandpapers. The polished, smooth planes ensured that the subsequent element distribution was tested on a plane. As shown in Fig. 4(a), the nano-sheets in the AFMO microspheres are compact. After calcination, the nano-sheets are transformed into nano-particles [Fig. 4(b)]. At the same time, some mesopores and macropores are spread throughout the AFLMOA microsphere, which is beneficial to the infiltration of the electrolyte. The results of element mapping [Fig. 4(a and b)] further confirm the existence of Al and F in the micro-spheres. The distribution data determined by line scanning of Ni, Co, Mn, Al, O, and F in AFMO and AFLMOA are shown in Fig. 4(c) and (e). For convenience, only the front half of the scanning lines is discussed, because the element distribution in the micro-sphere is symmetrical. To discuss the element change curves, there are two factors to be considered. They are the measured thickness and the elemental content, respectively. If the thickness at the edge of the half-sphere is less than the depth measured by the EDS, the increase of the measured thickness and the elemental content will lead to increasing signal strength of the elements. For AFMO [Fig. 4(c)], the content curves of Ni element show one steep slope in the distance from 0 to 1.3 μm and one gentle slope in the distance from 1.3 to 5.2 μm. The steep slope in the outer layer should be mainly caused by the change of test depth. In the distance from 1.3–5.2 μm, the gentle slope proves the concentration gradient distribution of element Ni. Similarly, Co, Mn, and Al also show an increasing trend in the distance from 0 to 1.3 μm. In contrast to Ni element, the concentration of Co remains stable in the distance from 1.3 to 5.2 μm. The change of Al concentration from 1.3 to 5.2 μm is not obvious. Most noteworthy is that the slope of the Al content curve from 0 to 1.3 μm is obviously less than that of Co. This indicates that Al is enriched in the outer layer. Surprisingly, the Mn content curve appears as a peak at 0.5 μm. This demonstrates that the outer layer from 0 to 1.3 μm has higher Mn content than the interior of the micro-spheres. This peculiar phenomenon is caused by the oxidation of Mn(OH)2. As confirmed by the XPS results, the Mn2+ on the surface of micro-spheres has been oxidated to Mn3+. Manganese oxides with Mn3+ (such as MnOOH and Mn2O3) have weak reactivity for ion exchange. In the interior of the micro-sphere, the Mn(OH)2 is not oxidized and could been partially exchanged. Therefore, the outer layer has higher Mn content than the interior of the micro-spheres. On the other hand, the concentration is low in the initial solid precursor Ni0.83Co0.10Mn0.07(OH)2. Under these conditions, the concentration gradient of Co is difficult to observe. The relative elemental intensity ratios across the AFMO sample particle are shown in Fig. 4(d); a similar method has been reported by Manthiram.28 The relative elemental Ni/Mn and Ni/Al intensity ratios across the AFMO sample particle increase from the surface to the center, while the Ni/Co intensity ratio remains almost constant from the surface to the center. This also indicates the formation of a Ni-rich phase in the center of the AFMO sample particle. In the final material, AFLMOA [Fig. 4(e) and (f)], the curves of element distribution are similar to those of its precursor, AFMO. Even though the elements are likely distributed uniformly after calcination, the gradients of Ni, Mn, and Al are maintained. A similar phenomenon was reported by Sun.26 In addition, the distribution of F always exhibits a similar shape to that of O (Fig. S3†), which indicates that the positions of O are partially substituted by F.
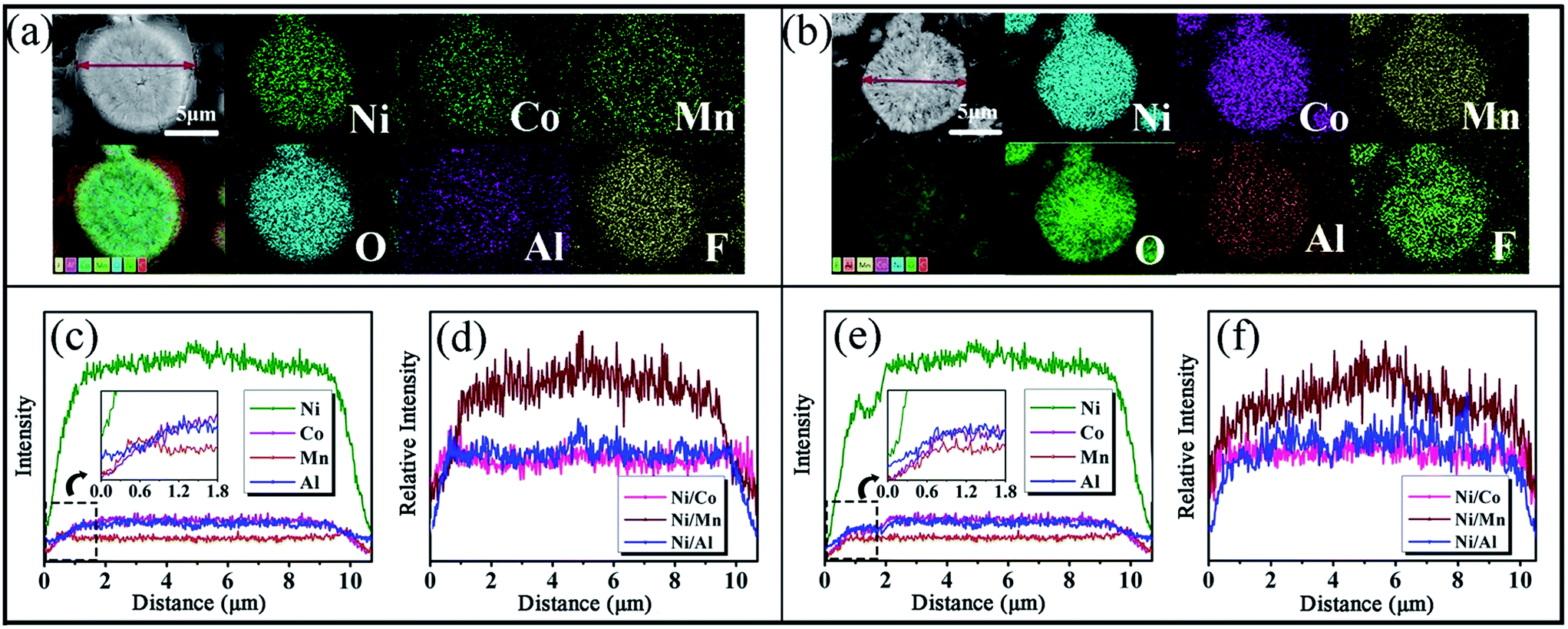 | ||
| Fig. 4 SEM and element mapping of cross sections of single micro-spheres of AFMO (a) and AFLMOA (b); the elemental (Ni, Co, Mn, Al) distribution line scanning of cross sections of single micro-spheres of AFMO (c) and AFLMOA (e); the EDS line scanning profile of the Ni content relative to the Co, Mn and Al content in single micro-spheres of AFMO (d) and AFLMOA (f). | ||
In addition, to show the results of the elemental (Ni, Co, Mn, Al) distribution more intuitively, the line scanning data have been transformed according to eqn (2)–(5). INi, ICo, IMn and IAl represent the intensities of Ni, Co, Mn and Al in the line scanning, respectively. The curves of IN, IC, IM and IA are shown in Fig. 5. The variation of the IN, IC, IM and IA curves could reflect the percentage content trend of Ni, Co, Mn and Al, respectively. In the distance from 0 to 1.0 μm, the curve of IN increases from the surface to the center. In contrast, the curves of IM and IA decline in the distance from 0 to 1.0 μm. The curve of IC remains almost level. These results show the distribution tendency of the relative amounts of elements (Ni, Co, Mn, Al). From the above-mentioned two data treatment methods, the gradient distribution of elements could be confirmed.
| IN = INi/(INi + ICo + IMn + IAl) | (2) |
| IC = ICo/(INi + ICo + IMn + IAl) | (3) |
| IM = IMn/(INi + ICo + IMn + IAl) | (4) |
| IA = IAl/(INi + ICo + IMn + IAl) | (5) |
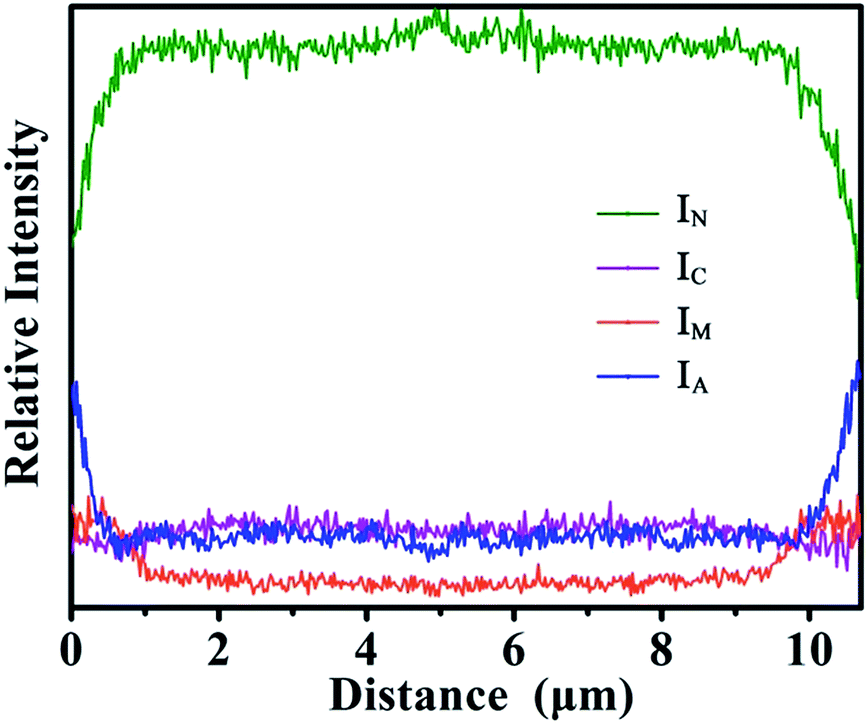 | ||
| Fig. 5 The curves of IN, IC, IM and IA in AFMO. | ||
In the hydrothermal environment at 120 to 180 °C, AlF3 exists as Al3+ and F−. Ion exchange initially occurs at the crystal surface. Then, the Al3+ and F− in the crystal surface diffuse inward gradually. In contrast the concentration gradient constructed by co-precipitation, the concentration gradient here is weaker. This phenomenon may be caused by the porous structure of the microspheres. The existence of holes in the outer layer contributes to the infiltration of solution in the ion exchange process, which shortens the ion exchange distance between the solution and the interior of the micro-spheres. Nevertheless, fluorine does not show an obvious graded distribution. This is caused by the fast dynamics of the F− exchange. The hydrated ionic radius of F− (3.52 Å)45 is smaller than that of Al3+ (4.80 Å), Ni2+ (4.04 Å), Co2+ (4.23 Å) and Mn2+ (4.38 Å), which benefits its migration in the AFMO particles.
The electrochemical performance of LMOA, AFLMOA, FLMOA and ALMOA was investigated as positive materials for Li-ion batteries. Charge and discharge curves of the 1st, 2nd, 5th, 10th, 30th and 40th cycles of the as-synthesized four materials are shown in Fig. 6(a–d). The materials LMOA, AFLMOA, FLMOA and ALMOA display first specific discharge capacity values of 148.7, 140.3, 157.9 and 166.4 mA h g−1, respectively. Meanwhile, the mean discharge voltages of LMOA, AFLMOA, FLMOA, and ALMOA are 3.75, 3.73, 3.78 and 3.79 V, respectively. FLMOA and ALMOA show higher specific discharge capacity and mean discharge voltage than LMOA in the first cycle. AFLMOA shows a slightly lower discharge specific capacity than LMOA, which may be ascribed to the cation mixing of the materials. After 40 cycles, their mean discharge voltage values decrease to 3.47, 3.54, 3.40 and 3.51 V, respectively. All materials show a decrease in mean discharge voltage. The decline of the mean discharge voltage is due to an increase in impedance, which is mainly caused by the appearance of rock salt phase (NiO, NaCl-type) on the surface of the cathode materials in the charge–discharge process.46 AFLMOA and ALMOA display the highest mean discharge voltage of the four materials at the 40th cycle. This is because the higher Al and Mn contents in the outer layer in the particles of AFLMOA can inhibit the exothermic reaction and decrease the impedance. Moreover, the doping of Al3+ can enhance the structural stability during the cycle.47
 | ||
| Fig. 6 The 1st, 2nd, 5th, 10th, 30th, and 40th charge–discharge curves of LMOA (a), AFLMOA (b), FLMOA (c), and ALMOA (d) at a current density of 20 mA g−1 at room temperature. Cyclic voltammogram curves [inset of (a–d)] of LMOA, AFLMOA, FLMOA and ALMOA at a scan rate of 0.1 mV s−1 between 3.0 and 4.5 V (vs. Li+/Li) at room temperature. The coulombic efficiency and cycle performance (e) of LMOA, AFLMOA, FLMOA, and ALMOA. The first charge–discharge curves (f) of AFLMOA and AFLMOO at a current density of 20 mA g−1 at room temperature. | ||
The CV curves are closely related with the structure of the materials. The Ni-based materials synthesized under O2 conditions have more orderly structures than the materials synthesized under air conditions. Therefore, the Ni-based materials synthesized under O2 conditions have sharp CV peaks. In contrast, detailed information of the changes in the metal oxidation state and structure cannot be reflected clearly in the CV curves of the Ni-based materials synthesized under air conditions.18,22 The CV curves of the initial and second cycles of all materials are presented in the inset of Fig. 6(a–d). In the first cycle, AFLMOA develops one pair of redox peaks at 4.16 and 3.48 V, respectively. The other materials, LMOA, FLMOA and ALMOA, do not evidently reveal anodic peaks because of intense polarization. The material LMOA has one broad cathodic peak in the first cycle. The cathodic peaks of FLMOA and ALMOA are more intense than that of LMOA, which is in agreement with the first specific discharge capacity results. In the second cycle, the obtained curve of LMOA displays a broad anodic peak (4.02 V) and a cathodic peak (3.25 V) with a peak potential separation of 770 mV. For AFLMOA, despite the negative shift in the anodic peak potential (3.98 V), no significant changes in the cathodic peak potential (3.45 V) were observed. The materials FLMOA and ALMOA both exhibit redox peaks at approximately 3.9 and 3.7 V. This value of the cathodic peak is slightly lower than the mean discharge voltage of FLMOA and ALMOA. The polarization of all materials decreases in the second cycle. This phenomenon should be attributed to the activation of the initial charge/discharge process.48 As expected, AFLMO, ALMOA and AFLMOA exhibit lower overpotential than LMOA. Especially, AFLMOA shows similar CV curves in the first and second cycles. This phenomenon indicates that AFLMOA is more stable than LMO, ALMOA, and FLMOA, which is consistent with the cyclic performance of charge–discharge. For the materials FLMOA and ALMOA, there are three reduction peaks in the second cycle, as shown in Fig. 6(c) and (d). This phenomenon reflects that the Ni-based materials synthesized in air conditions also undergo reduction reactions between Ni2+/Ni3+, Ni3+/Ni4+ and Co3+/Co4+ and three phase transitions in the process of discharge.
The cycle performance and coulombic efficiency of LMOA, AFLMOA, FLMOA and ALMOA are shown in Fig. 6(e). At the 1st to 10th cycles, the material LMOA performs serious capacity fading, which may be caused by the cation mixing and the high charge cut-off voltage. At high voltage (>4.2 V), the stability of LiMO2 (M = Ni, Co, and Mn) decreases, which would lead to poor cycle life.4 The capacity fading of AFLMOA, FLMOA and ALMOA at the first ten cycles is less than that of LMOA. After 40 cycles, the discharge capacity values of LMOA, AFLMOA, FLMOA, and ALMOA retain 53.87%, 74.70%, 57.51% and 58.49%, respectively. The materials FLMOA and ALMOA exhibit a slight increase in cycle performance. The material AFLMOA exhibits the highest cycling stability. The higher Al and Mn contents in the outer layer in the particles of AFLMOA can inhibit the exothermic reaction between the electrode and the electrolyte, while the inner Ni-enriched layer of the concentration gradient of AFLMOA delivers sufficient capacity. The graded distribution is beneficial to the electrochemical performance. On the other hand, the doping of Al3+ can hinder the change of the lattice parameters at the highly oxidized phase and enhance the structural stability.47 The substitution of F− can also stabilize the interface between the surface layers between the active particles and the electrolyte during cycling. The electrochemical performance results of a series of concentration gradient materials calcined in air with different ion exchange conditions are shown in Fig. S4 and S5.† The results indicate that the suitable gradient structures are beneficial for electrochemical performance. The initial discharge efficiencies of LMOA, AFLMOA, FLMOA, and ALMOA are 74.7%, 70.8%, 76.1%, and 78.7%, respectively. Their low initial discharge efficiencies should be attributed to the existence of cation mixing. Notably, except for the first cycle, the abovementioned four materials all show high coulombic efficiencies of about 100%.
Generally speaking, the discharge capacity of materials calcined in air is lower than that of materials calcined in O2 atmosphere in previous reports.48,49 This is because high Ni content materials calcined at air undergo severe cation mixing and low discharge capacity. For example, in a recent report,50 the material LiNiO2 calcined at air has a first specific discharge capacity of only 146 mA h g−1 in the first cycle at 0.1C. In comparison, the material AFLMOO was also calcined in O2 atmosphere at 700 °C to confirm the impact of atmosphere in the calcination process. As shown in Fig. 6(f), the material AFLMOO shows a first specific discharge capacity of 177.5 mA h g−1 at 0.1C in the first cycle with high coulombic efficiency (90.9%). Additional electrochemical data are shown in Fig. S6.† These results indicate that the low discharge capacities of the materials obtained in air are caused by the atmosphere in the process of calcination. Although the materials calcined in O2 have higher discharge capacity than the materials obtained in air, the cost of production of the materials obtained in O2 would also increase greatly. Therefore, the exploration of the materials in air with high performance is still significant.
DSC was performed to compare the thermal stabilities of LMO and AFLMOA in the 4.5 V charged state, as shown in Fig. 7. Compared with the material LMOA, the concentration gradient material AFLMOA showed a much lower amount of exothermic heat release around 220 °C. This indicates that the gradient structure and introduction of Al3+ and F− are beneficial to thermal stability.
 | ||
| Fig. 7 DSC profiles of LMOA and AFLMOA after charging at 4.5 V. | ||
Conclusions
In this work, a concentration gradient material, LiNi0.84Co0.10Mn0.04Al0.02O1.90F0.10, has been successfully synthesized by a new strategy. By ion exchange, Al3+ and F− have been introduced simultaneously into Ni0.83Co0.1Mn0.07(OH)2. In the final product, LiNi0.84Co0.10Mn0.04Al0.02O1.90F0.10, the distribution of the elements Al, Ni and Mn is a radial gradient. As a cathode in Li-ion batteries, the capacity retention after 40 cycles of the material LiNi0.84Co0.10Mn0.04Al0.02O1.90F0.10 calcined in air with low cost is 1.38 times that of LiNi0.83Co0.10Mn0.07O2 calcined in air. Meanwhile, LiNi0.83Co0.10Mn0.05Al0.02O2 and LiNi0.85Co0.10Mn0.05O1.97F0.03 obtained in air also show better cycling stability than the bare material LiNi0.83Co0.10Mn0.07O2; however, they show worse cycling stability than LiNi0.84Co0.10Mn0.04Al0.02O1.90F0.10. The better cycling stability of LiNi0.84Co0.10Mn0.04Al0.02O1.90F0.10 should be a benefit of its graded distribution. Although the materials calcined in air have slightly low discharge capacity, the production process is low cost and easy to achieve. On the other hand, the discharge capacity of LiNi0.84Co0.10Mn0.04Al0.02O1.90F0.10 obtained in O2 increases from 140.3 mA h g−1 to 177.5 mA h g−1. This result indicates that the low discharge capacities of the materials obtained in air are caused by the atmosphere during the process of calcination. This work provides a simple way to synthesize high Ni content concentration gradient cathode materials in air, which has potential applications in industry. Moreover, the simple strategy applied in this study could be used to design other materials, such as other metal hydroxides, metal oxides/sulfides, and lithium/sodium inset oxides.Acknowledgements
This work was supported by the Natural Science Foundation of China (No. U1407103), Henan Province (No. 15HASTIT003), Innovation Scientists and Technicians Troop Construction Projects of Henan Province and Zhengzhou University (No. 1421316035).Notes and references
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652 CrossRef CAS PubMed.
- J. B. Goodenough, Energy Environ. Sci., 2013, 7, 14 Search PubMed.
- V. Etacheri, R. Marom, R. Elazari, G. Salitra and D. Aurbach, Energy. Environ. Sci., 2011, 4, 3243 CAS.
- K. T. Lee, S. Jeong and J. Cho, Acc. Chem. Res., 2013, 46, 1161 CrossRef CAS PubMed.
- C. Wang, Z. Y. Guo, W. Shen, Q. J. Xu, H. M. Liu and Y. G. Wang, Adv. Funct. Mater., 2014, 24, 5511 CrossRef CAS.
- J. Lu, Q. Peng, W. Y. Wang, C. Y. Nan, L. H. Li and Y. D. Li, J. Am. Chem. Soc., 2013, 135, 1649 CrossRef CAS PubMed.
- J. Lu, C. Zhan, T. P. Wu, J. G. Wen, Y. Lei, A. J. Kropf, H. M. Wu, D. J. Miller, J. W. Elam, Y.-K. Sun, X. P. Qiu and K. Amine, Nat. Commun., 2014, 5, 5693 CrossRef CAS PubMed.
- M.-J. Lee, M. J. Noh, M.-H. Park, M. Jo, H. Kim, H. Nam and J. Cho, J. Mater. Chem. A, 2015, 3, 13453 CAS.
- H. Chen, J. A. Dawson and J. H. Harding, J. Mater. Chem. A, 2014, 2, 7988 CAS.
- J. Liu, K. P. Song, P. A. V. Aken, J. Maier and Y. Yu, Nano Lett., 2014, 14, 2597 CrossRef CAS PubMed.
- X. F. Li, J. Liu, X. B. Meng, Y. J. Tang, M. N. Banis, J. L. Yang, Y. H. Hu, R. Y. Li, M. Cai and X. L. Sun, J. Power Sources, 2014, 247, 57 CrossRef CAS.
- H. B. Shu, M. F. Chen, Y. Q. Fu, X. K. Yang, X. Yi, Y. S. Bai, Q. Q. Liang, Q. L. Wei, B. N. Hu, J. L. Tan, C. Wu, M. Zhou and X. Y. Wang, J. Power Sources, 2014, 252, 73 CrossRef CAS.
- M. Hu, Y. Tian, L. Su, J. Wei and Z. Zhou, ACS Appl. Mater. Interfaces, 2013, 5, 12185 CAS.
- M. Mladenov, R. Stoyanova, E. Zhecheva and S. Vassilev, Electrochem. Commun., 2001, 3, 410 CrossRef CAS.
- X. Zhou, Z. Dai, S. Liu, J. Bao and Y. G. Guo, Adv. Mater., 2014, 26, 3943 CrossRef CAS PubMed.
- H. Q. Li and H. S. Zhou, Chem. Commun., 2012, 48, 1201 RSC.
- Z. Q. Yi, L. X. Yuan, D. Sun, Z. Li, C. Wu, W. J. Yang, Y. W. Wen, B. Shan and Y. H. Huang, J. Mater. Chem. A, 2015, 3, 3059 CAS.
- L. W. Liang, K. Du, W. Lu, Z. D. Peng, Y. B. Cao and G. R. Hu, Electrochim. Acta, 2014, 146, 207 CrossRef CAS.
- Y. K. Sun, S. W. Cho, S. W. Lee, C. S. Yoon and K. Amine, J. Electrochem. Soc., 2007, 154, A168 CrossRef CAS.
- B. C. Park, H. B. Kim, S. T. Myung, K. Amine, I. Belharouak, S. M. Lee and Y. K. Sun, J. Power Sources, 2008, 178, 826 CrossRef CAS.
- T. Ohzuku, A. Ueda and M. Kouguchi, J. Electrochem. Soc., 1995, 142, 4033 CrossRef CAS.
- W. H. Chen, J. J. Zhao, Y. Y. Li, S. Li, C. C. Jin, C. C. Yang, X. M. Feng, J. M. Zhang and L. W. Mi, ChemElectroChem, 2014, 1, 601 CrossRef.
- L. J. Li, Z. Y. Chen, Q. B. Zhang, M. Xu, X. Zhou, H. L. Zhu and K. L. Zhang, J. Mater. Chem. A, 2015, 3, 894 CAS.
- Y. K. Sun, S. T. Myung, B. C. Park, J. Prakash, I. Belharouak and K. Amine, Nat. Mater., 2009, 8, 320 CrossRef CAS PubMed.
- P. Y. Hou, X. Q. Wang, D. W. Song, X. X. Shi, L. Q. Zhang, J. Guo and J. Zhang, J. Power Sources, 2014, 265, 174 CrossRef CAS.
- Y. K. Sun, Z. H. Chen, H. J. Noh, D. J. Lee, H. G. Jung, Y. Ren, S. Wang, C. S. Yoon, S. T. Myung and K. Amine, Nat. Mater., 2012, 11, 942 CrossRef CAS PubMed.
- W. C. Wen, S. H. Chen, Y. Q. Fu, X. Y. Wang and H. B. Shu, J. Power Sources, 2015, 274, 219 CrossRef CAS.
- J. Y. Liao and A. Manthiram, J. Power Sources, 2015, 282, 429 CrossRef CAS.
- D. H. Son, S. M. Hughes, Y. D. Yin and A. P. Alivisatos, Science, 2004, 306, 1009 CrossRef CAS PubMed.
- J. B. Rivest and P. K. Jain, Chem. Soc. Rev., 2013, 42, 89 RSC.
- W. T. Wei, L. W. Mi, Y. Gao, Z. Zheng, W. H. Chen and X. X. Guan, Chem. Mater., 2014, 26, 3418 CrossRef CAS.
- W. H. Chen, Y. F. Yang and H. X. Shao, J. Power Sources, 2011, 196, 488 CrossRef CAS.
- M. Saruyama, Y. G. So, K. Kimoto, S. Taguchi, Y. Kanemitsu and T. Teranishi, J. Am. Chem. Soc., 2011, 133, 17598 CrossRef CAS PubMed.
- A. Dianat, N. Seriani, M. Bobeth and G. Cuniberti, J. Mater. Chem. A., 2013, 1, 9273 CAS.
- G. Ming, Y. G. Li, H. B. Zhang, B. Zhang, W. Zhou, J. Feng, H. L. Wang, Y. Y. Liang, Z. J. Fan, J. Liu and H. J. Dai, Energy Environ. Sci., 2014, 7, 2025 Search PubMed.
- J. C. Huang, T. Lei, X. P. Wei, X. W. Liu, T. Liu, D. X. Cao, J. L. Yin and G. L. Wang, J. Power Sources, 2013, 232, 370 CrossRef CAS.
- P. Yue, Z. X. Wang, H. J. Guo, X. H. Xiong and X. H. Li, Electrochim. Acta, 2013, 92, 1 CrossRef CAS.
- W. E. Mofid, S. Ivanov, A. Konkin and A. Bund, J. Power Sources, 2014, 268, 414 CrossRef.
- A. N. Mansour, Surf. Sci. Spectra, 1994, 3, 239 CrossRef CAS.
- C. V. Schenck, J. G. Dillard and J. W. Murray, J. Colloid Interface Sci., 1983, 95, 398 CrossRef CAS.
- B. R. Strohmeier and D. M. Hercules, J. Phys. Chem., 1984, 88, 4922 CrossRef CAS.
- K. K. Lian, D. W. Kirk and S. J. Thorpe, J. Electrochem. Soc., 1995, 142, 3704 CrossRef CAS.
- A. Hess, E. Kemnitz, A. Lippitz, W. E. S. Unger and D. H. Menz, J. Catal., 1994, 148, 270 CrossRef.
- E. Kemnitz, A. Kohne, I. Grohmann, A. Lippitz and W. E. S. Unger, J. Catal., 1996, 159, 270 CrossRef CAS.
- A. G. Volkov, S. Paula and D. W. Deamer, Bioelectrochem. Bioenerg., 1997, 42, 153 CrossRef CAS.
- Y. Cho and J. Cho, J. Electrochem. Soc., 2010, 157, A625 CrossRef CAS.
- S.-W. Woo, S.-T. Myung, H. Bang, D.-W. Kim and Y.-K. Sun, Electrochim. Acta, 2009, 54, 3851 CrossRef CAS.
- C. Delmas, J. P. Pérès, A. Rougier, A. Demourgues, F. Weill, A. Chadwick, M. Broussely, F. Perton, Ph. Biensan and P. Willmann, J. Power Sources, 1997, 68, 120 CrossRef CAS.
- N. T. Wu, H. Wu, W. Yuan, S. J. Liu, J. Y. Liao and Y. Zhang, J. Mater. Chem. A, 2015, 3, 13648 CAS.
- K. Q. Ding, Y. B. Zhao, L. K. Liu, Y. Li, L. Liu, L. Wang, X. M. He and Z. H. Guo, Electrochim. Acta, 2015, 176, 240 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra03220f |
| This journal is © The Royal Society of Chemistry 2016 |