
Use of steel slag for CO2 capture under realistic calcium-looping conditions
Abstract
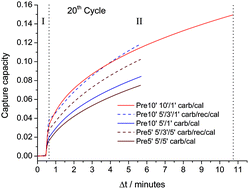
In this study, CaO derived from steel slag pretreated with diluted acetic acid has been tested as a dry sorbent for CO2 capture under realistic Ca-Looping (CaL) conditions, which necessarily implies calcination under high CO2 partial pressure and fast transitions between carbonation and calcination stages. The multicycle capture performance of the sorbent has been investigated by varying the precalcination time, carbonation/calcination residence times and with the introduction of a recarbonation stage. Results show that the sorbent can be regenerated in very short residence times at 900 °C under high CO2 partial pressure, thus reducing the calciner temperature by about 30–50 °C when compared to limestone. Although the introduction of a recarbonation stage to reactivate the sorbent, as suggested in previous studies for limestone, results in a slightly enhanced capture capacity, the sorbent performance can be significantly improved if the main role of the solid-state diffusion-controlled carbonation is not dismissed. Thus, a notable enhancement of the capture capacity is achieved when the carbonation residence time is prolonged beyond just a few minutes, which suggests a critical effect of solids residence time in the carbonator on the CO2 capture efficiency of the CaL process when integrated into a power plant.
 Please wait while we load your content...
Please wait while we load your content...

