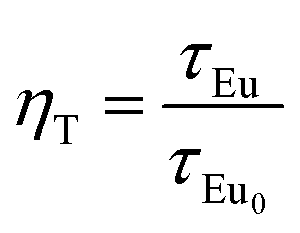DOI:
10.1039/C6RA03154D
(Paper)
RSC Adv., 2016,
6, 33990-33997
Eu2+ → Tb3+ → Eu3+ energy transfer in Ca6La2Na2(PO4)6F2:Eu, Tb phosphors
Received
3rd February 2016
, Accepted 28th March 2016
First published on 30th March 2016
Abstract
Novel phosphors Ca6La2Na2(PO4)6F2:0.10Eu2+/Eu3+, xTb3+ (x = 0, 0.05, 0.10, 0.20, 0.30, 0.40, 0.50) were prepared by a solid state reaction in a CO-reducing atmosphere. The fluorescence spectra of samples Ca6La2Na2(PO4)6F2:xEu2+ reveal that still a small number of Eu3+ ions are detected in the host. The Eu2+ → Tb3+ → Eu3+ energy transfer process in Ca6La2Na2(PO4)6F2:0.10Eu2+/Eu3+, xTb3+ phosphors was discussed in detail through the excitation, emission spectra and the fluorescence decay. As a result of Eu2+ sensitization, the relative emission intensity of Ca6La2Na2(PO4)6F2:0.10Eu2+/Eu3+, xTb3+ is enhanced under near-ultraviolet light excitation. Furthermore, tunable emission with a large color gamut can be obtained by changing the Tb3+ doping concentration. These results indicate that the Ca6La2Na2(PO4)6F2:Eu2+/Eu3+, Tb3+ phosphors will have potential use in n-UV chip pumped white LED devices.
1. Introduction
Nowadays, white light emitting diodes (w-LEDs) are been considered as the fourth-generation lighting source, and will take the place of conventional fluorescent lamps in the near future.1 For phosphor-converted w-LEDs, white light can be generated mainly in two ways: (1) fabricate a blue LED chip with the yellow-emitting phosphor Y3Al5O12:Ce3+; (2) fabricate an ultraviolet (UV) or near-UV (350–420 nm) LED chip with tri-color (red, green and blue) phosphors. The first way fabricated by blue-emitting chips coated with the yellow-emitting Y3Al5O12:Ce3+ garnet, have several drawbacks such as a high correlated color temperature (CCT) and a low color-rendering index (CRI). Accordingly, much attention has been paid to the second way in which tri-color phosphors are combined with ultraviolet (UV) or n-UV LEDs.2–4 As we know, the blue components of the n-UV based LEDs often come from the Ce3+ or Eu2+ doped phosphors,5–8 and Tb3+ and Eu3+ ions are usually used as green-emitting and red-emitting activators, because Tb3+ and Eu3+ ions can produce green and red emission due to 5D4 → 7FJ and 5D0 → 7FJ transitions, respectively.9,10 Furthermore, in Ce3+–Tb3+–Eu3+ and Eu2+–Tb3+–Eu3+ co-doped phosphors, Tb3+ ions can act as an energy transfer bridge like Ce3+/Eu2+ → Tb3+ → Eu3+ between Ce3+/Eu2+ and Eu3+ ions. It is well known that Ce3+ and Eu2+ ions are excellent sensitizers, however, the energy transfer efficiency for Ce3+/Eu2+ → Eu3+ is usually low. This is largely due to the metal–metal charge transfer (MMCT), in which excited charge-transfer state Ce4+–Eu2+ is easily occur.11,12 So it is a good way to enhance Eu3+ emission by energy transfer like Ce3+/Eu2+ → Tb3+ → Eu3+ ions. In present, some Ce3+–Tb3+–Eu3+ and Eu2+–Tb3+–Eu3+ co-doped phosphors were reported, and it is a novel way to obtain bright phosphors and control the emission color.13–18
It is well known that the compounds with the apatite structure are considered as effective host lattices for luminescent materials. Among these compounds, the fluoro-apatite compounds with general formula M10−2xLnxNax(PO4)6F2 (M = Ca, Sr, Ba; Ln = rare earths) are studied widely for luminescent materials.19–21 In our previous work, RE3+ (Ce3+, Tb3+, Eu3+, Dy3+, Tm3+ and Sm3+)-doped M10−2xLnxNax(PO4)6F2 (M = Ca, Sr, Ba; Ln = rare earths) phosphors were studied.22–27 In this work, novel phosphors Ca6La2Na2(PO4)6F2:Eu2+/Eu3+ (CLPF:Eu) and Ca6La2Na2(PO4)6F2:Eu2+/Eu3+,Tb3+ (CLPF:Eu, Tb) were synthesized, and the detailed photoluminescence properties were investigated for their potential application in n-UV based LEDs.
2. Experimental
Eu2+/Eu3+ doped phosphors Ca6La2Na2(PO4)6F2:xEu2+/Eu3+ (x = 0.01, 0.04, 0.07, 0.10, 0.13, 0.16, 0.20) and Eu2+/Eu3+–Tb3+ co-doped Ca6La2Na2(PO4)6F2 phosphors Ca6La2Na2(PO4)6F2:0.10Eu, xTb (x = 0, 0.05, 0.10, 0.20, 0.30, 0.40, 0.50) were synthesized by a high-temperature solid-state reaction. The starting materials are analytical grade CaCO3, NH4H2PO4, NH4F, Na2CO3 and rare earth oxides (La2O3, Eu2O3, 99.95%; Tb4O7, 99.99% purity). The raw materials were carefully weighed stoichiometrically and ground in an agate mortar. After mixing and thorough grinding, the mixtures were heated at 1050 °C for 4 h in a CO-reduced atmosphere, by covering the highly pure carbon grains on the inner crucible. The final products were cooled to room temperature by switching off the muffle furnace and ground again into white powder.
The phase purity and structure of the final products were characterized by a powder X-ray diffraction (XRD) analysis using Cu Kα radiation (λ = 1.5405 Å, 40 kV, 30 mA) on a PANalytilal X'pert Powder X-Ray Diffractometer at room temperature (RT). The photoluminescence properties were measured on a HITACHI F7000 fluorescence spectrometer equipped with a 450 W xenon lamp as the excitation source. The luminescence decay spectra were measured by a FLS 920 steady-state spectrometer equipped with a fluorescence lifetime spectrometer, and a 150 W nF900 ns flash lamp and a 60 W μF flash lamp were used as the flash-light source, respectively. All the measurements were performed at room temperature (RT).
3. Results and discussion
3.1 Phase characterization
As apaties, the compounds M10−2xLnxNax(PO4)6F2 (M = Ca, Sr, Ba; Ln = rare earths) have general structure of the fluoapatites with P63/m space group.27 Two cationic M2+ (Ln3+) sites can be found in these compounds. Site (1) are at the Wyckoff 4f positions with C3 point symmetry which is surrounded by nine oxygen anions, while the other site (2) are at the Wyckoff 6h positions with Cs point symmetry which is surrounded by six oxygen anions plus one F− anion. Though different amounts of cations M2+ and Ln3+ will bring different distortion of the tetrahedra structure, significant changes in the structure do not occur. So the JCPDS card of Ca5(PO4)3F can be used to identify the phase purity of Ca6La2Na2(PO4)6F2 in previous work.23,27
The phase purities of the as-prepared samples were measured by X-ray diffraction (XRD) at room temperature. Fig. 1 shows the XRD patterns of typical samples CLPF:xEu (a–c), CLPF:0.10Eu, xTb (d–f) and the Joint Committee on Powder Diffraction Standards (JCPDS) card file (no 71-0881) of Ca10(PO4)6F2 as a reference. It can be seen that the diffraction patterns of these samples are in good agreement with the standard JCPDS card, and no other peaks or impurities are detected. Hence, it can be concluded that the dopant Eu2+, Eu3+ and Tb3+ ions are completely incorporated into the host lattice without making significant changes to the crystal structure.
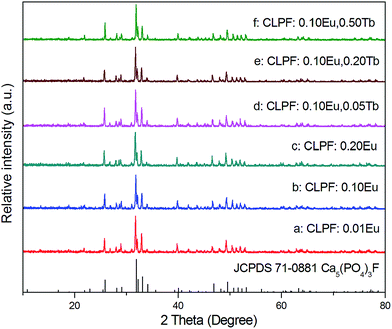 |
| | Fig. 1 XRD patterns of samples CLPF:xEu (a–c) and CLPF:0.10Eu, xTb (d–f). | |
3.2 Luminescent properties of CLPF:xEu
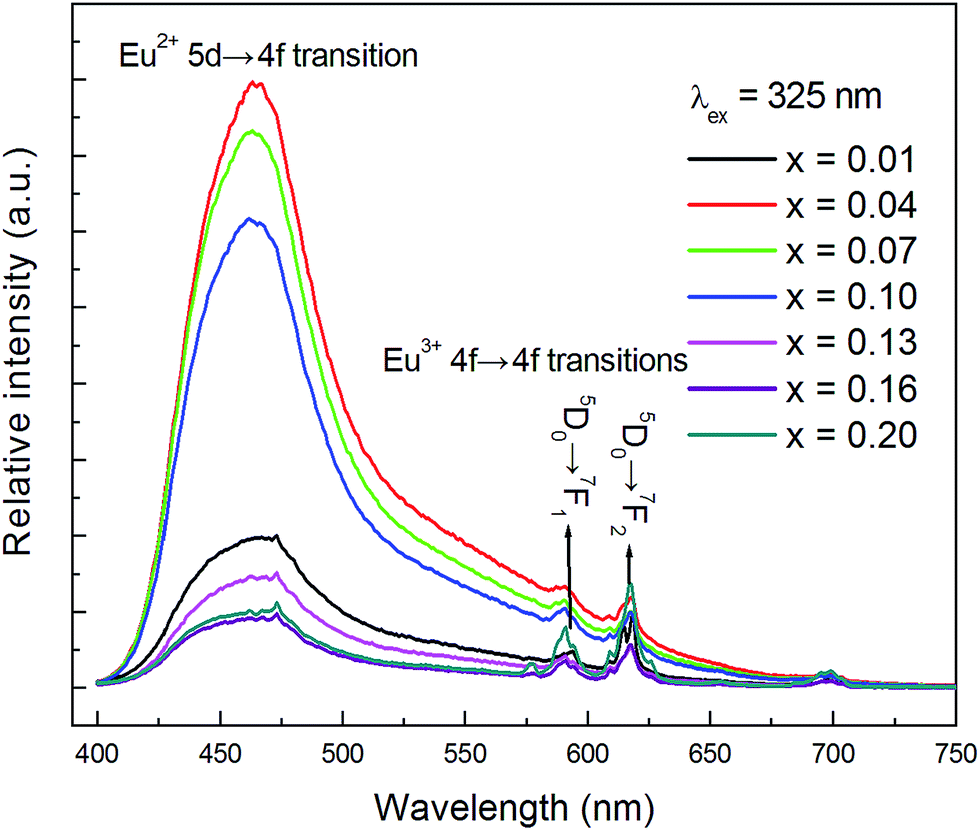
The PL spectra (λex = 325 nm) of CLPF:xEu (x = 0.01, 0.04, 0.07, 0.10, 0.13, 0.16, 0.20) phosphors are presented in Fig. 2. It can be seen that all these phosphors exhibit a broad blue emission band with a peak at around 465 nm, which corresponds to the 4f65d1 → 4f7 allowed transition of Eu2+. It is clear that the PL intensity increases greatly with Eu concentration increasing first, reaches a maximum at a low Eu concentration (x = 0.04), and then decreases with increasing Eu doping concentrations, which should be due to the concentration quenching effect. Furthermore, it is evident that Eu3+ is still detected in all these phosphors, especially for samples with high doping concentration. There are two small narrow emission lines with peaks centred at 593 nm and 618 nm in the spectra, which correspond to the 5D0 → 7F1 and 5D0 → 7F2 transition of Eu3+, respectively.23
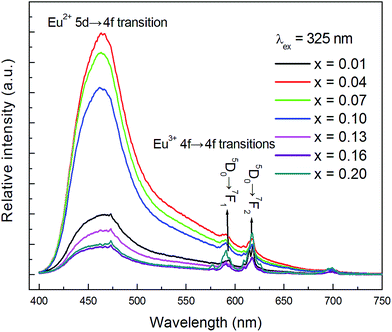 |
| | Fig. 2 PL spectra of samples CLPF:xEu (x = 0.01, 0.04, 0.07, 0.10, 0.13, 0.16, 0.20) under 325 nm excitation. | |
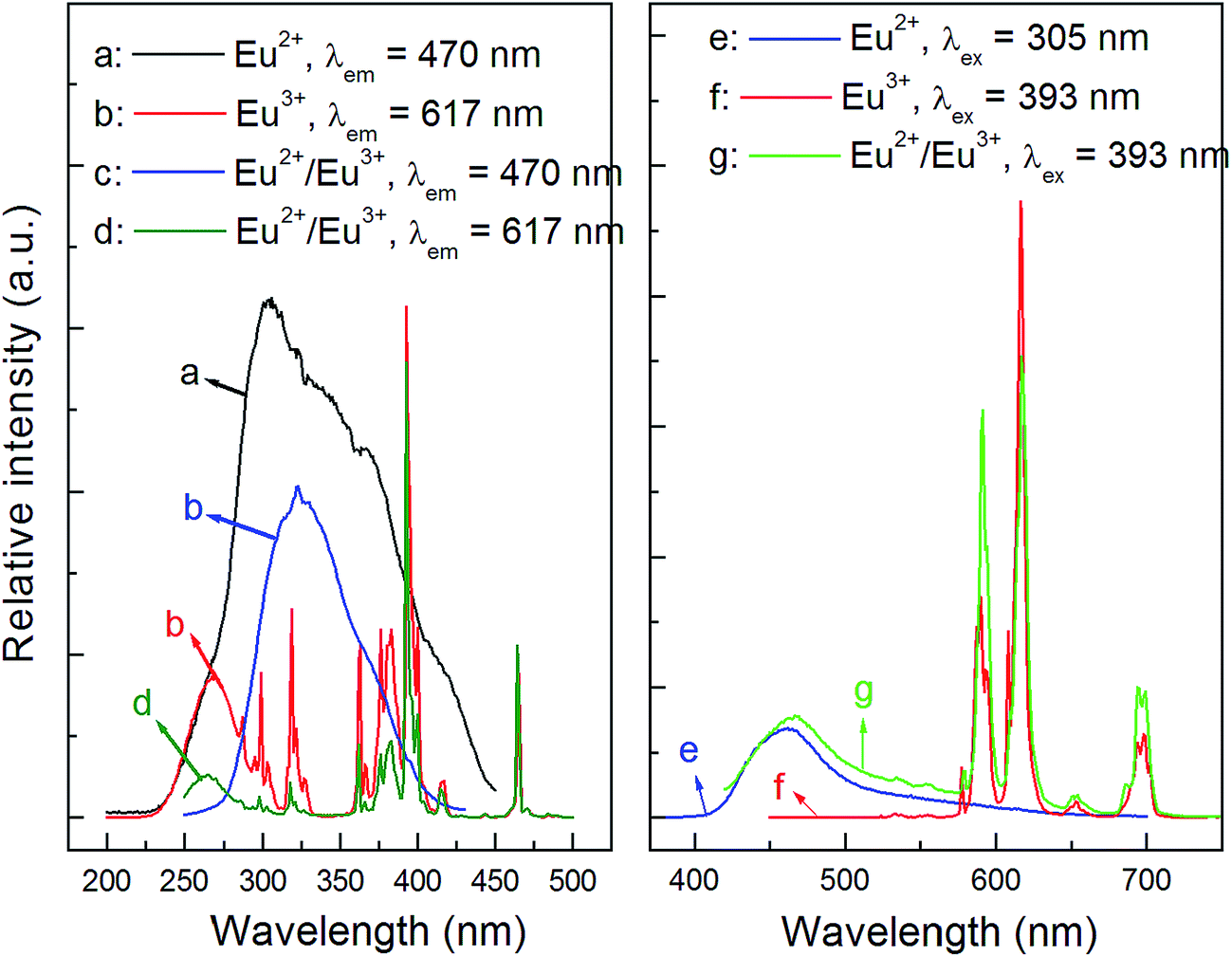
As is known to all, the detailed electronic structure of Eu2+ is [Xe]4f75d0 while it is [Xe]4f6 for Eu3+. Thus, the allowed 4f75d0 → 4f65d1 transition of Eu2+ would occur when it is excited, and show a broad emission band, while Eu3+ ions emit narrow lines owing to the f–f transitions. According to the observation of Eu valence state in phosphors in previous reports,16,28–32 we propose that both Eu2+ and Eu3+ can exist stably in the CLPF host. To prove it, a Eu3+ single-doped CLPF phosphor CLPF:0.10Eu3+ was prepared in air and a Eu2+ single-doped phosphor CLPF:0.10Eu2+ was prepared in H2-reducing atmosphere for comparing. The PLE and PL spectra of samples CLPF:0.10Eu3+, CLPF:0.10Eu2+/Eu3+ and CLPF:0.10Eu2+ are shown in Fig. 3. As shown in Fig. 3(a), a broad excitation band centered at 305 nm originated from 4f75d0 → 4f65d1 of Eu2+ can be observed. The PLE spectrum (Fig. 3(b)) of Eu3+ single-doped phosphor CLPF:Eu3+ consists of a broad excitation band in the 200–300 nm wavelength region and a series of excitation peaks from 300 to 500 nm, which are attributed to charge transfer band and f–f transitions of Eu3+ ions. The PLE spectrum (Fig. 3(c) and (d)) profile of CLPF:0.10Eu2+/Eu3+ are nearly identical to that of Eu2+ singly-doped phosphor CLPF:0.10Eu2+ and Eu3+ singly-doped CLPF:0.10Eu3+ except for intensity when they are monitored at 470 and 594 nm, respectively. It confirms that Eu2+ and Eu3+ ions should coexist in the CLPF host.
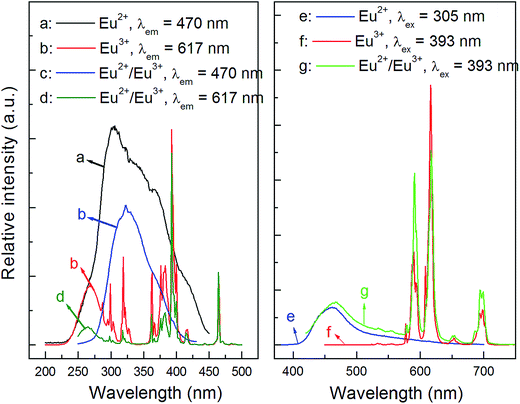 |
| | Fig. 3 PLE and PL spectra of samples CLPF:0.10Eu2+ ((a): λem = 470 nm; (e): λex = 305 nm), CLPF:0.10Eu3+ ((b): λem = 617 nm; (f): λex = 393 nm) and CLPF:0.10Eu2+/Eu3+ ((c): λem = 470 nm; (d): λem = 617 nm; (g): λex = 393 nm). | |
The emission spectrum (Fig. 3(e)) of sample CLPF:0.10Eu2+ under 305 nm excitation consists of a broad emission band in the blue region with a peak centred at 465 nm. It can be seen clearly that the emission peak position is almost coincident with that of CLPF:Eu phosphors as in Fig. 2. And the emission spectrum (Fig. 3(f)) of CLPF:0.10Eu3+ under 393 nm excitation shows series of narrow emission lines with peaks centred at 591 nm, 617 nm and 700 nm, which correspond to the 5D0 → 7F1, 5D0 → 7F2 and 5D0 → 7F4 transition of Eu3+, respectively. The emission peak position of Eu3+ ions is the same as that in our previous work.23 As seen Fig. 3(g), it is not difficult to conclude that the emission spectrum of sample CLPF:0.10Eu2+/Eu3+ under 393 nm excitation consists of both Eu2+ emission band and Eu3+ emission lines.
3.3 Eu2+ → Tb3+ → Eu3+ energy transfer in CLPF:Eu, Tb
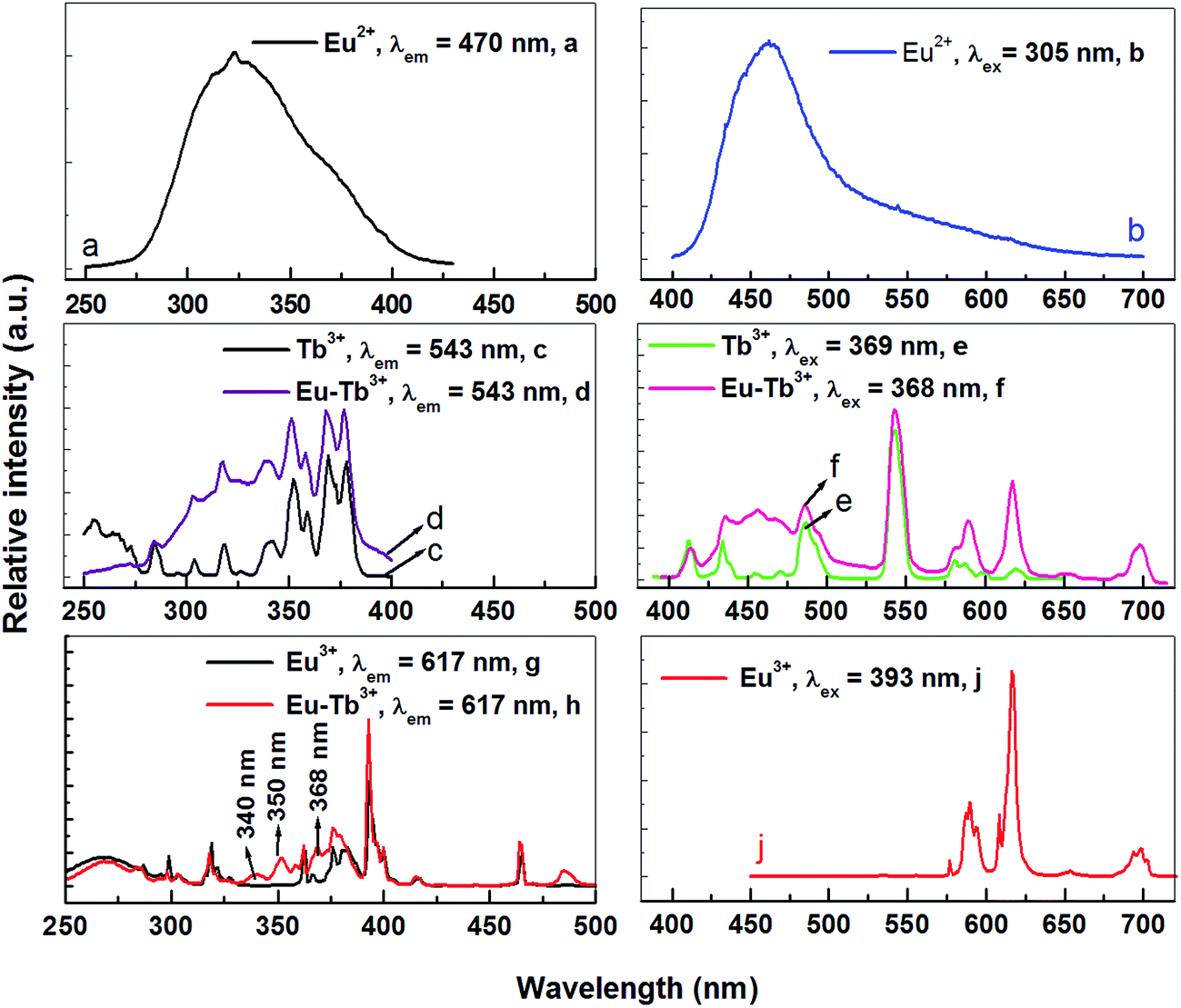
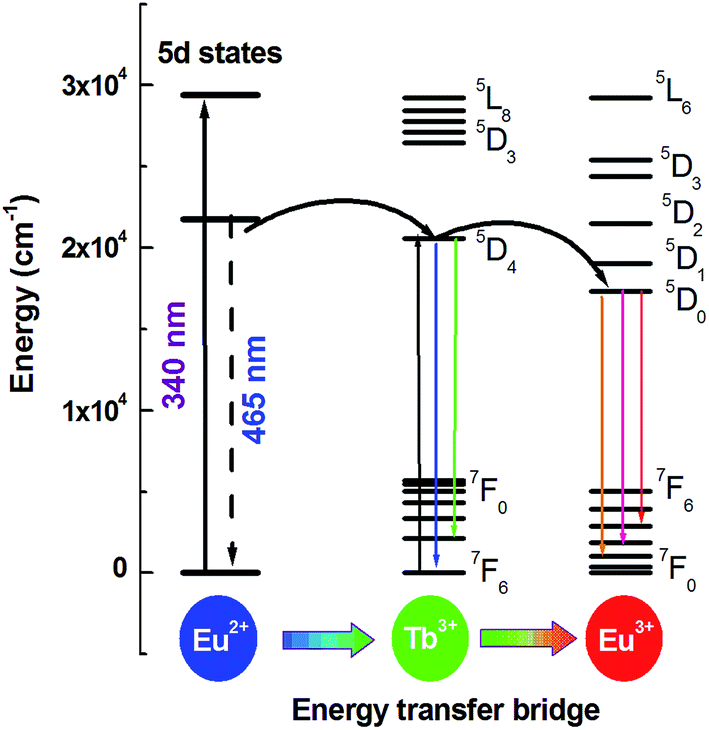
As mentioned above, Eu3+ ions are hard to be directly sensitized by Ce3+ or Eu2+ ions ascribed to the existence of metal–metal charge transfer (MMCT), which will quenche the luminescence of the sensitizer.11,12 However, Eu2+ → Tb3+ and Tb3+ → Eu3+ can be found in many hosts. So can Tb3+ ion be used as a terbium bridge to realize a Eu2+ → Tb3+ → Eu3+ energy transfer in CLPF host? Based on this idea, we prepared series of samples CLPF:0.10Eu2+/Eu3+, xTb3+ (x = 0.05, 0.10, 0.20, 0.30, 0.40 and 0.50), which is shortened as CLPF:0.10Eu, xTb. The PL and PLE spectra of phosphors CLPF:0.10Eu2+, CLPF:0.10Eu3+, CLPF:0.50Tb3+ and CLPF:0.10Eu, 0.10Tb are shown in Fig. 4. By comparing the PL spectra of these single-doped and co-doped, it is easy to find characteristic Eu2+ emission band and Eu3+, Tb3+ emission lines in the emission spectrum of sample CLPF:0.10Eu, 0.10Tb under 368 nm excitation (curve f). By comparing the PLE spectra of samples CLPF:0.10Tb3+ and CLPF:0.10Eu, 0.10Tb (curve c and d), it can be found that the excitation intensity of CLPF:0.10Eu, 0.10Tb phosphor is enhanced when Eu is introduced. It is clear that Eu2+ → Tb3+ energy transfer occurs in this case. The PLE spectra of samples CLPF:0.10Eu3+ and CLPF:0.10Eu, 0.10Tb (curve g and h) show that Tb3+ excitation lines (340, 350 and 368 nm) are observed by monitoring Eu3+ emission at 617 nm, which indicates that Tb3+ → Eu3+ energy transfer is realized. The detail energy transfer process is shown in Fig. 5. When Eu2+ ions are excited by n-UV light, part of energy can be transfer to the 5D4 level of Tb3+ ions from Eu2+, and then the energy is further transferred to the 5D0 level of Eu3+ ions, and finally Eu3+ emission intensity is enhanced. Therefore, the Tb3+ ions act as both activators and sensitizers at the same time in CLPF:Eu, Tb phosphors, which we call it as energy transfer bridge. Further discuss about this energy transfer model can be found in latter part by PL spectra and decay curves.
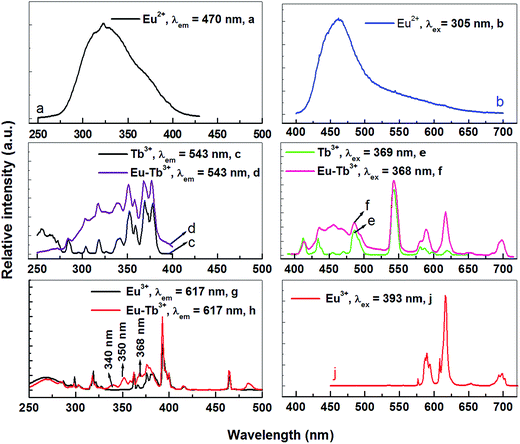 |
| | Fig. 4 PL and PLE spectra of CLPF:0.10Eu2+, CLPF:0.10Eu3+, CLPF:0.50Tb3+ and CLPF:0.10Eu, 0.10Tb phosphors. | |
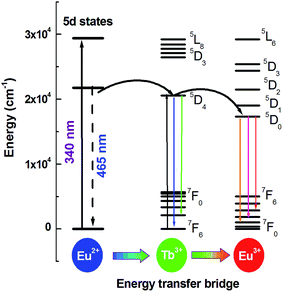 |
| | Fig. 5 Schematic diagram of energy transfer Eu2+ → Tb3+ → Eu3+ in CLPF:Eu, Tb. | |
Fig. 6 shows emission spectra of CLPF:0.10Eu, 0.10Tb phosphor under different light excitation. Except for curves a and f excited by 260 and 400 nm, in which only Eu3+ emission peaks are observable, the Eu2+ emission band (∼470 nm), Tb3+ emission lines (∼487, 543 nm) and the Eu3+ emission lines (∼591, 617 and 700 nm) can be found from other emission spectrum curves. Still the relative intensities of Eu2+, Eu3+ and Tb3+ in these curves are different. This is not surprising, because different wavelength may directly excite different luminescent centre ions, and the energy transfer behaviors are also different. Interestingly, 305 nm light can excite neither Tb3+ nor Eu3+ ions, and 340, 368 nm light can hardly excite Eu3+ ions, which we can conclude from the excitation spectra of CLPF:0.50Tb3+ and CLPF:0.10Eu (curve c and g in Fig. 4). However, Tb3+ and Eu3+ emission peaks can be clearly seen in curve b, c and d excited by 305, 340, 368 nm light. This result further indicates that energy transfer process Eu2+ → Tb3+ → Eu3+ occurs. In order to further discuss Eu2+ → Tb3+ and Tb3+ → Eu3+ energy transfer, the emission spectra under 340 and 305 nm excitation for samples CLPF:0.10Eu2+/Eu3+, xTb3+ with different Tb3+ doping concentration are investigated latter.
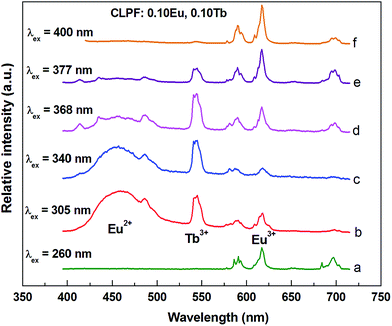 |
| | Fig. 6 PL spectra of sample CLPF:0.10Eu, 0.10Tb excited by different wavelength. | |
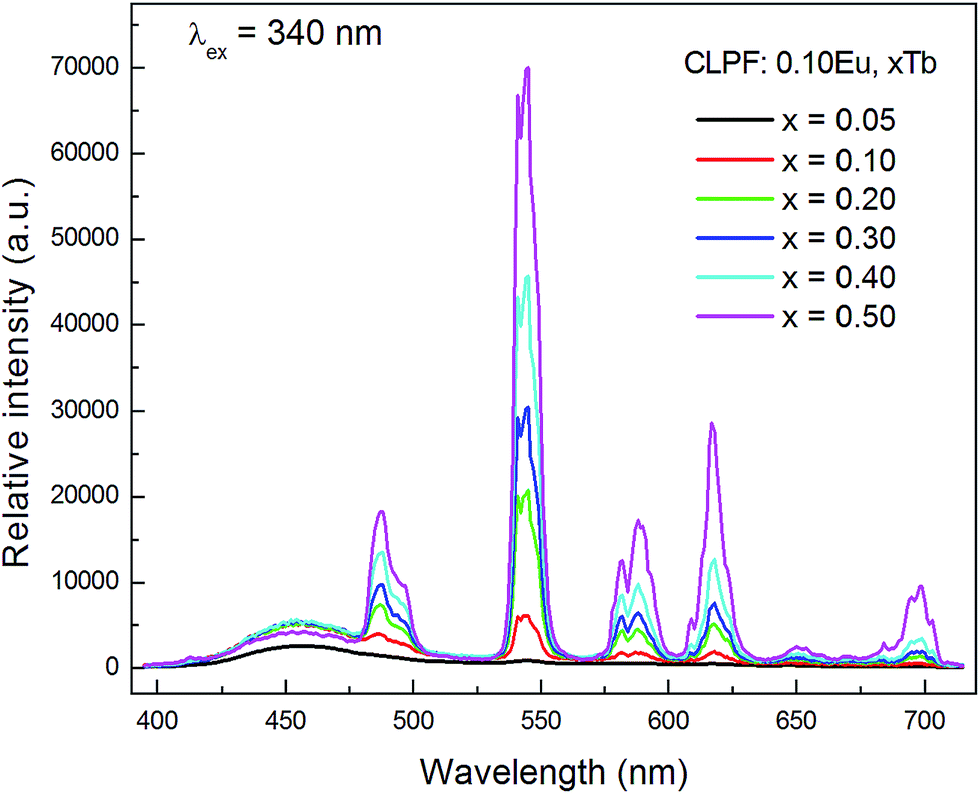
Fig. 7 shows the concentration dependence of PL spectra for the samples CLPF:0.10Eu, xTb (x = 0.05, 0.10, 0.20, 0.30, 0.40 and 0.50) under 340 nm excitation. The characteristic Eu2+, Tb3+ and Eu3+ emission bands and peaks can be found from all these PL spectra. As discussed above, Tb3+ and Eu3+ strong emission peaks in this case is due to Eu2+ → Tb3+ → Eu3+ energy transfer. The emission intensity of Eu2+, Tb3+ and Eu3+ is plotted in the inset of Fig. 8. It can be seen that the intensity of Eu2+ does not change much, and has the optimum emission intensity with doping Tb3+ concentration at x = 0.10. The Eu3+ and Tb3+ relative intensities increase evidently with Tb3+ concentration increasing. It is interesting that the Eu3+ emission intensity of CLPF:0.10Eu, 0.50Tb is enhanced over fifty times that of CLPF:0.10Eu due to the Eu2+ → Tb3+ → Eu3+ energy transfer. As compared, the PL spectra of the samples CLPF:0.10Eu, xTb under 305 nm excitation are also measured and presented in Fig. 9. It is evident that Eu2+ relative intensities are more obvious in this case than that in Fig. 7. It is ascribed to the fact that 305 nm light can excite Eu2+ more effective than 340 nm.
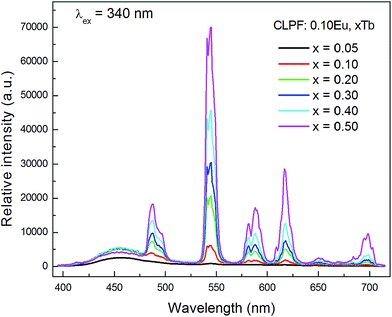 |
| | Fig. 7 PL spectra for 340 nm excitation of samples CLPF:0.10Eu, xTb (x = 0.05, 0.10, 0.20, 0.30, 0.40 and 0.50). | |
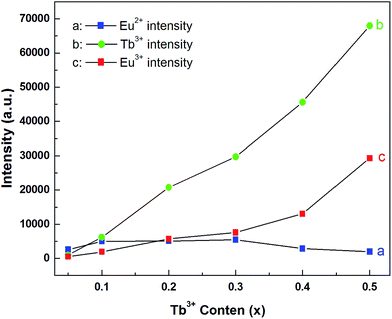 |
| | Fig. 8 Eu2+, Eu3+ and Tb3+ emission intensities in dependent of Tb3+ concentrations in CLPF:0.10Eu, xTb (x = 0.05, 0.10, 0.20, 0.30, 0.40 and 0.50). | |
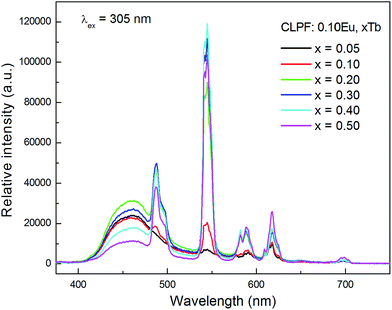 |
| | Fig. 9 PL spectra of samples CLPF:0.10Eu, xTb (x = 0.05, 0.10, 0.20, 0.30, 0.40 and 0.50) under 305 nm excitation. | |
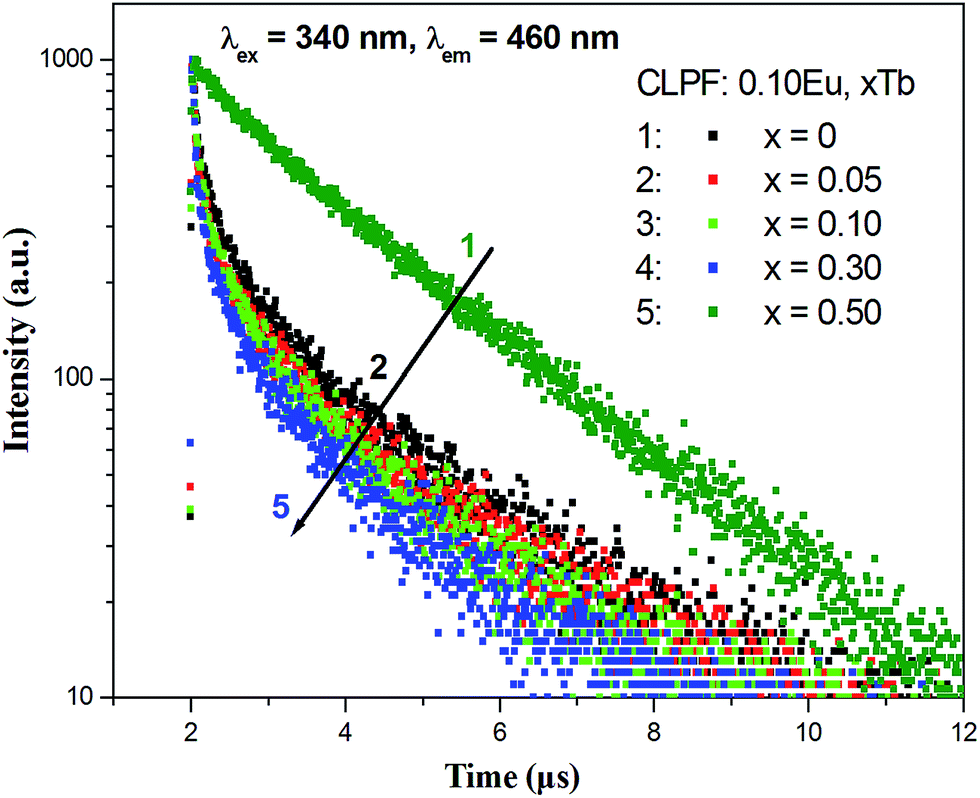
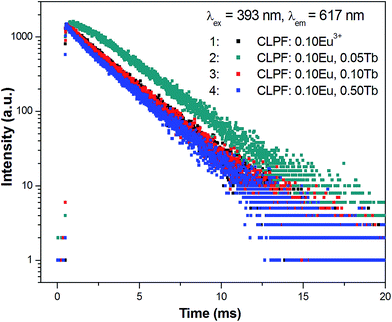
Fig. 10 presents the room temperature decay curves of Eu2+ in CLPF:0.10Eu, xTb (x = 0, 0.05, 0.10, 0.30 and 0.50). The lifetime of Eu2+ can be fitted by a single-order exponential equation in the absence of Tb3+, and the lifetime value is calculated to be 2.04 μs. When Tb3+ is introduced, the lifetime decreases evidently and becomes a second-order exponential decay. With Tb3+ concentration increasing, the lifetime of Eu2+ gradually decrease. This result clearly indicates that an energy transfer process between Eu2+ and Tb3+ exists in CLPF:0.10Eu, xTb phosphors. Similar results have been discussed extensively in previous work.16 The energy transfer efficiency from Eu2+ to Tb3+ can be calculated according to the following equation:16,33
| |
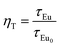 | (1) |
where
τEu0 and
τEu are the decay time of Eu
2+ in the absence and presence of acceptor Tb
3+, respectively. The calculated results of
ηT are listed in
Table 1, which shows that the energy transfer efficiency from Eu
2+ to Tb
3+ change increases from 77.9% to 86.3% with Tb
3+ doping concentration increasing.
 |
| | Fig. 10 Decay curves of Eu2+ in CLPF:0.10Eu, xTb (x = 0, 0.05, 0.10, 0.30 and 0.50). | |
Table 1 The lifetime values of Eu2+ in CLPF:0.10Eu, xTb and the energy transfer efficiency (ηT) from Eu2+ to Tb3+
| No. |
x value |
TEu (μs) |
ηT (%) |
| 1 |
0 |
2.04 |
NA |
| 2 |
0.05 |
0.45 |
77.9 |
| 3 |
0.10 |
0.32 |
84.3 |
| 4 |
0.30 |
0.30 |
85.3 |
| 5 |
0.50 |
0.28 |
86.3 |
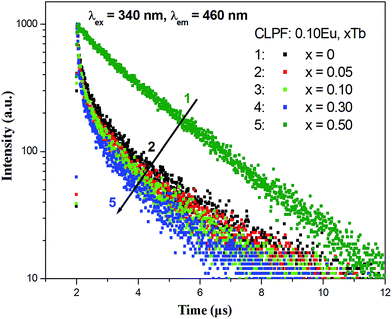
Fig. 11 gives the decay curves for the Eu3+ emission at 617 nm in CLPF:0.10Eu, xTb (x = 0.05, 0.10 and 0.50) and CLPF:0.10Eu3+ phosphors. When the Tb3+ concentration is low (x = 0.05), the decay curves show a clear rise immediately after the excitation pulse which indicates feeding of the Eu3+ excited states. This further confirms the energy transfer process form Tb3+ to Eu3+. As the Tb3+ concentration increases, the rise process gets faster and faster, and the decay of the Eu3+ become single exponential, which are in good agreement with the Eu3+ decay of in Eu3+ singly-doped phosphor CLPF:0.10Eu3+.
 |
| | Fig. 11 Decay curves of Eu3+ in CLPF:0.10Eu, xTb (x = 0.05, 0.10 and 0.50) and CLPF:0.10Eu3+. | |
3.4 CIE chromaticity coordinates and diagram for CLPF:Eu, Tb
In order to investigate the application in n-UV phosphor converted LEDs, n-UV lights (368 and 377 nm) are also chosen to excite the CLPF:Eu, xTb phosphors, and the emission spectra are shown in Fig. 12. It can be seen that the relative intensities of Eu2+, Eu3+ and Tb3+ vary with the increasing of Tb3+ concentration. By comparing the panels a and b in Fig. 12, it is not difficult to find out the difference: Eu2+ emission in panel b is weaker than that in panel a. It is because 368 nm light can excite Eu2+ ions more efficiency than 377 nm. Based on these emission spectra, we can easily predict that the emitting color of CLPF:Eu, xTb phosphors is tunable under n-UV light excitation. The CIE chromaticity diagram and CIE chromaticity coordinates for the CLPF:Eu, xTb under 368 nm excitation are shown in Table 2 and Fig. 13. As seen in Fig. 13, It can be seen that the emitting color of the CLPF:Eu, xTb phosphors under 368 nm excitation can be modulated from blue to white, to yellow-green, and even to yellow. Similarly, under 377 nm excitation, the CIE coordinates of phosphors with low Tb3+ doping concentrations (x = 0.05 and 0.10) locate at white light region, and change to yellow region gradually with Tb3+ doping concentrations increasing. Thus, the emitting colors can be tunable in a large color gamut by changing the Tb3+ doping concentration. Furthermore, the white-light emission can be obtained in a single host, and the CIE coordinates can be further modulated to be closer to the white point (x = 0.33, y = 0.33) by varying the Tb3+ doping concentration.
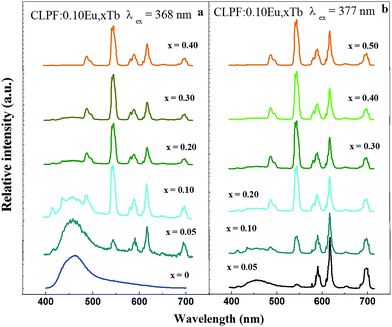 |
| | Fig. 12 PL spectra of CLPF:Eu, xTb phosphors under 368 and 377 nm excitation. | |
Table 2 CIE chromaticity coordinates for CLPF:Eu, xTb under 368 and 377 nm excitation
| No. |
x value |
CIE coordinates (x, y) (λex = 368 nm) |
CIE coordinates (x, y) (λex = 377 nm) |
| 1 |
0 |
(0.193, 0.193) |
NA |
| 2 |
0.05 |
(0.258, 0.229) |
(0.380, 0.273) |
| 3 |
0.10 |
(0.284, 0.301) |
(0.381, 0.324) |
| 4 |
0.20 |
(0.332, 0.428) |
(0.382, 0.433) |
| 5 |
0.30 |
(0.345, 0.457) |
(0.383, 0.460) |
| 6 |
0.40 |
(0.399, 0.508) |
(0.395, 0.480) |
| 7 |
0.50 |
NA |
(0.425, 0.498) |
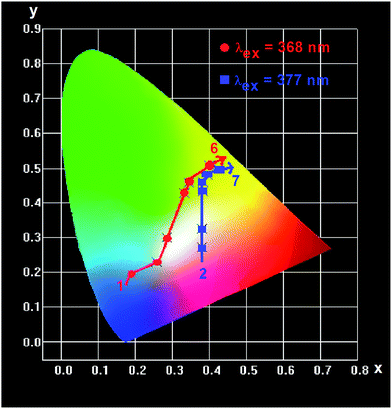 |
| | Fig. 13 CIE chromaticity diagram for CLPF:Eu, xTb phosphors under 368 (red dot) and 377 nm (blue dot) excitation. | |
4. Conclusions
Eu3+ ions were detected in the so-called CLPF:xEu2+, CLPF:0.10Eu2+, xTb3+ phosphors which were prepared by a solid state reaction in a CO reducing atmosphere. In CLPF:0.10Eu, xTb phosphors, Tb3+ ions act as a Tb energy transfer bridge, and the Eu2+ → Tb3+ → Eu3+ energy transfer is realized. The energy transfer efficiency of Eu2+ → Tb3+ can reach 86.3% in CLPF:0.10Eu, 0.50Tb. The emission intensity n-UV light excitation can be enhanced obviously because of Eu2+ sensitizing. The emission color of CLPF:0.10Eu, xTb have a large color gamut by changing the Tb3+ doping concentration. Under 368 and 377 nm excitation, the emission color changes continuously from blue to white and eventually to yellow region as the concentration of the Tb3+ increases. Hence, CLPF:0.10Eu, Tb phosphors can act as a single-component white-light phosphor for wLEDs.
Acknowledgements
The work is financially supported by National Natural Science Foundation of China (Grant no. 21401165, 11404283), Natural Science Foundation of Guangdong Province (Grant no. 2014A030307040, 2014A030307028), Training Program for Excellent Youth Teachers in Universities of Guangdong Province (No. YQ2015110), Overseas Scholarship Program for Elite Young and Middle-aged Teachers of Lingnan Normal University, and the Fundamental Research Funds for the Central University (No. 21614334).
References
- Y. H. Song, G. S. Han, S. R. Mang, M. K. Jung, H. S. Jung and D. H. Yoon, J. Mater. Chem. C, 2015, 3, 235 RSC.
- Y. S. Tang, S. F. Hu, W. C. Ke, C. C. Lin, N. C. Bagkar and R. S. Liu, Appl. Phys. Lett., 2008, 93, 131114 CrossRef.
- Y. Y. Zhang, Z. G. Xia, H. K. Liu, Z. Y. Wang and M. L. Li, Chem. Phys. Lett., 2014, 593, 189 CrossRef CAS.
- M. B. Xie, L. H. Zeng, T. L. Ye, X. Yang, X. M. Zhu, S. Y. Peng and L. Lei, Mater. Res. Express, 2014, 1, 035005 CrossRef.
- H. S. Jang, Y. H. Won and D. Y. Jeon, Appl. Phys. B, 2009, 95, 715 CrossRef CAS.
- Y. Q. Li, G. With and H. T. Hintzen, J. Mater. Chem., 2005, 15, 4492 RSC.
- M. M. Jiao, Y. C. Jia, W. Lü, W. Z. Lv, Q. Zhao, B. Q. Shao and H. P. You, J. Mater. Chem. C, 2014, 2, 4304 RSC.
- C. L. Zhao, Z. G. Xia and M. L. Li, RSC Adv., 2014, 4, 33114 RSC.
- K. Li, J. Fan, M. M Shang, H. Z. Lian and J. Lin, J. Mater. Chem. C, 2015, 3, 9989 RSC.
- C. H. Huang and T. M. Chen, J. Phys. Chem. C, 2011, 115, 2349 CAS.
- G. Blasse and A. Bril, J. Chem. Phys., 1967, 47, 1920 CrossRef CAS.
- G. Blasse, Phys. Status Solidi A, 1983, 75, K41 CrossRef CAS.
- S. C. Xu, P. L. Li, Z. J. Wang, T. Li, Q. Y. Bai, J. Sun and Z. P. Yang, J. Mater. Chem. C, 2015, 3, 9112 RSC.
- X. Chen, J. F. Zhao, L. P. Yu, C. Y. Rong, C. Z. Li and S. X. Lian, J. Lumin., 2011, 131, 2697 CrossRef CAS.
- D. W. Wen, J. X. Shi, M. M. Wu and Q. Su, ACS Appl. Mater. Interfaces, 2014, 6, 10792 CAS.
- Z. G. Xia, J. Q. Zhuang, H. K. Liu and L. B. Liao, J. Phys. D: Appl. Phys., 2012, 45, 015302 CrossRef.
- Z. G. Xia, J. Q. Zhuang, A. Meijerink and X. P. Jing, Dalton Trans., 2013, 42, 6327 RSC.
- M. P. Saradhi, S. Boudin, U. V. Varadaraju and B. Raveau, J. Solid State Chem., 2010, 183, 2496 CrossRef CAS.
- I. Mayer and S. Cohen, J. Solid State Chem., 1983, 48, 17 CrossRef CAS.
- I. Mayer, R. S. Roth and W. E. Brown, J. Solid State Chem., 1974, 11, 33 CrossRef CAS.
- M. Toumi, L. Smiri-Dogguy and A. Bulou, J. Solid State Chem., 2000, 149, 308 CrossRef CAS.
- M. B. Xie, H. B. Liang, Y. Tao, Y. Huang and Q. Su, Inorg. Chem., 2010, 49, 11317 CrossRef CAS PubMed.
- M. B. Xie and R. K. Pan, J. Alloys Compd., 2013, 551, 48 CrossRef CAS.
- M. B. Xie, H. B. Liang, B. Han, W. P. Chen, Q. Su, Y. Huang, Z. H. Gao and Y. Tao, Opt. Lett., 2009, 34, 3466 CrossRef CAS PubMed.
- M. B. Xie, Y. Huang, Y. Tao, H. B. Liang and Q. Su, J. Electrochem. Soc., 2010, 157, J401 CrossRef CAS.
- M. B. Xie, H. B. Liang, Q. Su, Y. Huang, Z. H. Gao and Y. Tao, Electrochem. Solid-State Lett., 2010, 13, J140 CrossRef CAS.
- M. B. Xie, S. L. Ou, H. B. Liang, D. J. Hou, Y. Huang, Z. H. Gao and Y. Tao, Mater. Chem. Phys., 2013, 142, 339 CrossRef CAS.
- Z. W. Pei, Q. H. Zeng and Q. Su, J. Phys. Chem. Solids, 2000, 61, 9 CrossRef CAS.
- A. Dobrowolska and E. Zych, J. Phys. Chem. C, 2012, 116, 25493 CAS.
- A. Baran, S. Mahlik, M. Grinberg, P. Cai, S. I. Kim and H. J. Seo, J. Phys.: Condens. Matter, 2014, 26, 385401 CrossRef CAS PubMed.
- S. C. Xu, P. L. Li, Z. J. Wang, T. Li, Q. Y. Bai, J. Sun and Z. P. Yang, J. Mater. Chem. C, 2015, 3, 9112 RSC.
- X. Chen, J. F. Zhao, L. P. Yu, C. Y. Rong, C. Z. Li and S. X. Lian, J. Lumin., 2011, 131, 2697 CrossRef CAS.
- M. B. Xie, D. Y. Li, R. K. Pan, X. P. Zhou and G. X. Zhu, RSC Adv., 2015, 5, 22856 RSC.
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.